
An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List

A Review of Liposomes as a Drug Delivery System: Current Status of Approved Products, Regulatory Environments, and Future Perspectives
Liposomes have been considered promising and versatile drug vesicles. Compared with traditional drug delivery systems, liposomes exhibit better properties, including site-targeting, sustained or controlled release, protection of drugs from degradation and clearance, superior therapeutic effects, and lower toxic side effects. Given these merits, several liposomal drug products have been successfully approved and used in clinics over the last couple of decades. In this review, the liposomal drug products approved by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) are discussed. Based on the published approval package in the FDA and European public assessment report (EPAR) in EMA, the critical chemistry information and mature pharmaceutical technologies applied in the marketed liposomal products, including the lipid excipient, manufacturing methods, nanosizing technique, drug loading methods, as well as critical quality attributions (CQAs) of products, are introduced. Additionally, the current regulatory guidance and future perspectives related to liposomal products are summarized. This knowledge can be used for research and development of the liposomal drug candidates under various pipelines, including the laboratory bench, pilot plant, and commercial manufacturing.
1. Introduction
Liposomes are self-assembled (phospho)lipid-based drug vesicles that form a bilayer (uni-lamellar) and/or a concentric series of multiple bilayers (multilamellar) enclosing a central aqueous compartment [ 1 ]. The size of liposomes ranges from 30 nm to the micrometer scale, with the phospholipidbilayer being 4–5 nm thick [ 2 ]. The field of liposomology was launched by the British scientist Alec Bangham and colleagues at Babraham Cambridge in the mid-1960s [ 3 ], and they first published the structure of liposomesin 1964 [ 4 ]. Since then, liposomes have been widely investigated as delivery vehicles for small molecular drugs, protein, nucleic acid, and imaging agents [ 5 , 6 , 7 , 8 , 9 ]. Different administration routes, such as parenteral, pulmonary, oral, transdermal, ophthalmic, and nasal routes, have been developed to improve therapeutic efficacy and patient compliance [ 10 , 11 , 12 , 13 , 14 ]. In addition, liposomes have been widely applied in the fields of food [ 15 ] and cosmetics [ 16 ].
As drug vehicles, liposomes exhibit outstanding properties, such as protecting the encapsulated substances from physiological degradation [ 17 ], extending the half-life of the drug, controlling the release of drug molecule s [ 18 ], and excellent biocompatibility and safety. Furthermore, liposomes can selectively deliver their payload to the diseased site through passive and/or active targeting, thus decreasing the systemic side-effect, elevating the maximum-tolerated dose, and improving therapeutic benefits [ 19 , 20 ].
Unlike normal tissue with tight intracellular junctions (2–6 nm) between endothelial cells [ 21 ], abnormal tissues such as a solid tumor or inflammatory site have highly porous capillaries (100 nm–2 µm depending upon the size and type of tumor tissue [ 22 ]). Liposomes can cross over the discontinuous neovasculature and be passively accumulated and detained at the abnormal tissues, which is called the enhanced permeability and retention (EPR) effect. Actively targeting employs specific interactions between the ligands and receptors on the surface of liposomes and tumor cells, respectively. Tumor cells may overexpress specific receptors, such as vascular endothelial growth factor (VEGF), epidermal growth factor (EGF), folic acid (FA), integrin, CD44 (a cell surface glycoprotein), CD13, and prostate-specific membrane antigen [ 23 ]. According to these receptors, specific ligands, such as antibody [ 24 ], nuclear acid (e.g., aptamers [ 25 ]), protein (e.g., transferrin [ 26 ]), peptides (e.g., iRGD [ 27 ], iNGR [ 28 ]), small molecules (folic acid [ 29 ]), and carbohydrates (e.g., dextran, mannose, and galactose [ 30 ], targeting macrophages) were proposed for the surface modification of liposomes.
Besides the specific medicines, liposomes stand as an excellent technique for drug delivery. However, only 14 types of liposomal products are available on the market, which means the advantages of liposomes have not been fully exploited. Therefore, in this review, we summarized the knowledge about commercial liposomal products approved by the FDA and EMA. Attention is paid to the composition and manufacturing technologies adopted in commercial products. In addition, the CQAs of liposomes, the current regulatory environment, and future perspectives are introduced. The purpose of this review is to provide important reference information to accelerate the development of liposomes.
2. The Marketed Liposomal Products
We searched the approved drug database published on the website of the FDA and EMA, and found that 14 types of liposomal products have been authorized ( Table 1 ). It should be noted that this list excludes generics, lipid complexes (e.g., Abelcet, Amphotec, and Onpattro), and nationally authorized liposomal products in Europe. Doxil(Doxorubicin HCl liposome injection) was the first liposomal product approved by the FDA in 1995. Among these marketed products, 43% of products were approved before the year 2000, and 57% of products were approved before the year 2010. The therapeutic area mainly focuses on cancer therapy but also involves other areas, such as infection, anesthesia, vaccine, lung disease, and photodynamic therapy. The dosage forms are mainly focused on sterile suspension and lyophilization powder. The administration routes include intravenous infusion, intramuscular and intrathecal injection, epidural, local infiltration, and oral inhalation.
Summary of liposomal products approved by FDA and EMA.
This list is only for liposomal forms approved by FDA and EMA, excludes generics (e.g., doxorubicin hydrochloride (liposomal), lipid complexes (e.g., Abelcet, Amphotec, and Onpattro), and also excludes the nationally authorized liposomal products in Europe. Abbreviations: intravenous infusion (IV), intramuscular injection (IM), intrathecal injection (IT), lyophylization (Lyo), muramyl tripeptide phosphatidyl ethanolamine (MTP-PE).
3. Structures and Main Components of Liposomes
3.1. structures of liposomes.
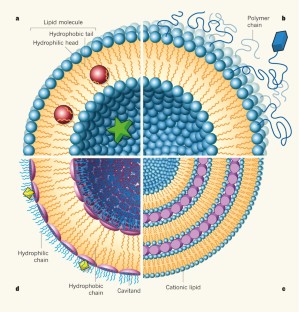
Liposomes can be classified as unilamellar vesicles (ULVs), oligolamellar vesicles (OLVs), multilamellarvesicles (MLVs), and multivesicular liposomes (MVLs) depending on the compartment structure and lamellarity ( Figure 1 ) [ 31 ]. OLVs and MLVs show anonion-like structure but present 2–5 and >5 concentric lipid bilayers, respectively. Different from MLVs, MVLs include hundreds of non-concentric aqueous chambers bounded by a single bilayer lipid membrane and display a honeycomb-like structure [ 32 ]. Based on the particle size, ULVs can be further divided into small unilamellar vesicles (SUVs, 30–100 nm), large unilamellar vesicles (LUVs, >100 nm), and giant unilamellar vesicles (GUVs, >1000 nm) [ 33 ]. Different size range of ULVs was reported, i.e., SUVs with a size of less than 200 nm and LUVs with a size of 200–500 nm [ 34 ].

Categories and structures of liposomal drug delivery system. ( a ) Structural illustration of liposome composition. The size of a typical phospholipid bilayer is 4.5 nm, which is much smaller than the one of the inner aqueous core; ( b ) Classification of liposomal vesicles according to their lamellarity/compartment and particle size; ( c ) The size and lamellarity of different types of liposomes; ( d , e ) The cryo-transmission electron microscopy of Doxil [ 35 ] and Vyxeos [ 36 ]; ( f , g ) The electron micrographs of DepoFoam TM particles with a typical diameter of 1–100 μm (e.g., DepoCyt) and MLVs with a typical diameter of 0.2–5 μm (e.g., Mepact) [ 37 ].
The particle size and the structures of the commercial products are concluded in Table 2 . Most of the current commercial products are SUVs (e.g., Doxil ( Figure 1 d) because of the long circulation time and ability to passively target the diseased site. Arikaye (amikacin liposome inhalation suspension) is considered an LUV because of its large particle size (200–300 nm). Vyxeos (daunorubicin: cytarabine liposome for injection) is a bilamellar liposome system ( Figure 1 e), which is generated during the first drug cytarabine loading procedure. The mechanism of internal lamella formation is explained as a thermodynamic response of the lipid bilayer to decrease the surface area-to-volume ratio of the liposomes caused by water egress in response to an external osmotic challenge [ 36 ]. Myocet (liposomal doxorubicin) and Mepact (liposomal mifamurtide powder for concentrate for dispersion for infusion) ( Figure 1 g) are MLVs. The abundant lamellae provide a large space for the encapsulation of lipophilic compounds [ 38 ].
The particle size, structure, and lipid components of the commercial liposomal products.
Abbreviations: fully hydrogenated soy phosphatidylcholine (HSPC), egg phosphatidylcholine (EPC), distearoylphosphatidylcholine (DSPC), dioleoyl phosphatidylcholine (DOPC), dierucoyl phosphatidylcholine (DEPC), palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), dipalmitoyl phosphatidylcholine (DPPC), dimyristoylphosphatidylcholine (DMPC), dipalmitoylphosphatidylglycerol (DPPG), distearoylphosphatidylglycerol (DSPG), dioleoyl phosphatidylserine(DOPS), dioleoylphosphatidylserine (OOPS), cholesterol (Chol), sphingomyelin (SM), N-(carbonyl-methoxypolyethlyeneglycol-2000)-distearolyphosphatidylethanolamine (MPEG-2000-DSPE).
There are four products with micron diameters, i.e., Mepact, DepoCyt (cytarabine liposome suspension), DepoDur (morphine sulfate extended-release liposome injection), and Exparel (bupivacaine liposome injectable suspension). Mepactis a sterile and lyophilized cake, and will form multilamellar liposomes with a particle size of 2.0–3.5 µm after reconstituted with 0.9% saline solution. This size is ideal for recognition and phagocytosis by monocytes/mocrophages, and triggers the macrophages and the immunomodulatory effects for cancer therapy. DepoCyt, DepoDur, and Exparel are manufactured by the same DepoFoam technique. The MVLs structure ( Figure 1 f) can load a large volume of drug–aqueous solution because of the numerous chambers and provide sustained release due to the erosion/degradation of liposomes and diffusion of drug molecules [ 39 , 40 ].
3.2. Main Components of Liposomes
Table 2 shows that glycerolphospholipid (GP), sphingomyelin (SM), and cholesterol (Chol) are the basic components used in the marketed products. GP contains glycerol, which links a pair of hydrophobic fatty acid chains and a hydrophilic polar head group [ 48 ]. The types of fatty acid and polar heads are described in Figure 2 a. Under the physiological pH, different head groups provide liposomes with negative (PA, PS, PG, and cardiolipin) or neutral (PC and PE) charges [ 49 ].

( a ) Structural illustration of glycerolphospholipid. R1 and R2 can be saturated or unsaturated fatty acids, such as decanoic acid, lauric acid, palmitic acid, oleic acid, myristic acid, stearic acid, and erucic acid. R3 can be phosphatidylcholine (PC), phosphatidyl ethanolamine (PE), phosphatidyl serine (PS), phosphatidyl inositols (PI), phosphatidic acid (PA), phosphatidylglycerol (PG), and cardiolipin; ( b ) Structure of sphingomyelin. ( c ) Structure of cholesterol.
Negatively charged DSPG used in AmBisome (ambisome liposome for injection) can interact with the positively charged amine group of AmpB to form a stable ionic complex [ 50 ], while DSPG used in Vyxeos minimize liposome aggregation by a strong Coulombic repulsive force [ 51 ]. DSPC used in DaunoXome (daunorubicin citrate liposome injection), Onivyde (irinotecan liposome injection), and Vyxeos is a neutral and synthetic lipid with well-defined fatty acid composition (two molecules of stearic acid), high purity, and a relatively high phase transition (T m of 55 °C).
EPC is adopted as an excipient in Myocet and Visudyne (verteporfin powder for solution for infusion). EPC is a naturally sourced phospholipid (NPL) purified from egg yolk. Compared to semi-synthetic and synthetic lipids, NPL exhibit a low production cost, but a broad transition temperature, problematic to obtain completely identical NPL and potential batch variation of liposomes [ 52 ]. In addition, the unsaturated fatty acid of EPC leads to a low phase transition temperature of −15~−5 °C [ 53 ], indicating the liposome bilayer is in a disorder and drug “leaky” state in the body temperature. Myocet, composed of EPC, is unstable in blood, and most drugs are released after 24 h [ 54 ]. Visudyne composed by EPC and DMPC also exhibits less stability in the presence of serum. The verteporfin rapidly transfers from the disordered liposome membrane and associates with the plasma lipoproteins, then reaches higher levels in the neovasculature since low-density lipoprotein (LDL) receptors are abundant in neovascular tissue [ 55 , 56 ].
MethoxyPEG (Mw 2000 Da), covalently attached to DSPE (MPEG-DSPE) used in Doxil and Onivyde, provides “stealth” and sterically stabilized liposomes. The molecular weight of PEG and the mole percentage of PEG-DSPE in lipid composition play important roles on the bilayer packing, circulation time, and thermodynamic stability. The high molecular weight of PEG (>2000 Da) grafted to the lipid headgroup exhibits repulsive forces from the liposome surface, as well as protects liposomes from binding serum proteins and avoids further clearance by the mononuclear phagocytic system (MPS), but also decreases the interaction and endocytosis of liposomes by targeted cells [ 57 ]. The low molecular weight of PEG (<750 Da) shows an insignificant steric stabilization effect [ 58 ]. Additionally, the highest biological stability of liposomes can be obtained when the concentration of PEG-DSPE is 7 ± 2 mol% in the lipid assemblies, and 5 mol% of PEG-lipid conjugates as a typical concentration have been used in vivo (e.g., Doxil) [ 58 , 59 ]. In the case of the concentration of PEG-DSPE below 4 mol%, the PEG chains shows “mushroom” configuration with a thickness about 3.5 nm. As the increased concentration of 4–8 mol%, the configuration of PEG chains transforms to “brush” with a thickness of 4.5–10 nm [ 58 , 60 ]. Further increasing the molar ratio, micelles are formed instead of liposome assembling.
DepoCyte, DepoDur, and Exparel have special structures and similar lipid components. A minimum of two types of lipids in the formulation are required for MVLs formation, the amphipathic lipid and the neutral lipid (e.g., diglycerides, triglycerides, vegetable oil) [ 61 ]. DOPC and DEPC are amphipathic zwitterionic phospholipids that form the walls of honeycomb-like chambers. DPPG with a negative charge prevents the MVLs from aggregation [ 62 ]. Neutral lipids (e.g., triolein and triglycerides) act as a hydrophobic space filler at bilayer intersection points and stabilize these membrane junctions [ 63 ]. Without the neutral lipids, conventional ULVs or MLVs will be formed instead of MVLs. The amount of neutral lipids used in formulation decides the capture volume and encapsulation efficiency of MVLs [ 63 ].
GPs play a key role in formulation since they affect the biophysical properties of liposomes (e.g., drug encapsulation, stability, and drug release) and further influence the pharmacokinetic behavior and pharmacodynamics in vivo [ 64 ]. The length, symmetry, inter- and intra-molecular interactions, branching, and unsaturation degree of hydrocarbon chains decide the thickness and fluidity of the bilayer, phase transition temperature, and drug release rate [ 49 , 65 ]. In brief, the longer hydrocarbon chain could induce a tighter membrane packing and increase drug retention, whereas the higher degree of unsaturation or branching of the hydrocarbon chain could result in looser membrane packaging, which is probably caused by the preferential interaction of cholesterol with saturated phospholipids in comparison with unsaturated ones [ 66 , 67 ].
Sphingomyelin (SM) ( Figure 2 b) has a similar structure toa glycerolphospholipid, except that glycerol is replaced by sphingosine [ 68 ]. Marqibo (vincristine sulfate liposome injection) uses SM to form the bilayer membrane, which significantly decreases the lipid hydrolysis in an acidic environment and prompts the stability of liposomes. An acid environment (pH 2.0–4.0) is routinely used for producing a transmembrane pH gradient for active drug loading. Under the condition of 37 °C and pH 2.0, the rate of hydrolysis for liposomes was approximately 100-fold slower in SM/Chol (55/45, mol/mol) liposomes than in DSPC/Chol liposomes [ 69 ]. In addition, liposomes with SM/Chol showed optimal pharmacokinetic properties, i.e., increased circulation time and enhanced delivery of the drug to target tissues [ 70 ].
Cholesterol (Chol) ( Figure 2 c) is another main component of the liposome bilayer and can be used in almost all commercial products ( Table 2 ). The addition of Chol can promote the packing of lipid chains and bilayer formation [ 71 ], modulate the fluidity/rigidity of membrane [ 72 ], and further affect the drug release [ 73 , 74 ], stability of liposomes [ 75 ], and the kinetics of exocytosis [ 76 ]. For the product of Shingrix (herpes zoster vaccine, contains glycoprotein E antigen and AS01 B liposomal adjuvant system), Chol can avoid the hydrolysis of QS21 (one of the immunoenhancers in the AS01 B adjuvant system) at a ratio of 2:1 (Chol: QS21, w / w ) [ 77 ]. For the product of AmBisome, Chol reduces the toxicity of the liposomal formulation compared with the non-sterol one [ 41 ]. The effect of Chol on the bilayer’s properties is concentration-dependent. It was reported that [ 71 ] the low (2.5 mol%) and high (>30 mol%) concentrations of Chol showed little effect on the properties of the lipid bilayer. The “condensing effect” or “ordering effect” of Chol with the content of 5 < Chol mol% < 30 led to a gradual increase in particle size from 220 nm to 472 nm, a decrease in the fluidity of membrane, and a decrease in the release of drug. Besides Chol, other sterols, such as progesterone, ergosterol, and lanosterol, with a similar structure to Chol were also investigated to modulate the membrane rigidity and stability [ 74 , 78 ].
4. Manufacturing Process
Various preparation methods of liposomes have been developed. The potential manufacturing processes of the marketed liposomal products summarized based on the related patents and publications are shown in Figure 3 [ 79 ]. The commonly used manufacturing processes include the thin-film hydration, ethanol injection, and double emulsion methods. The processes routinely include (1) the preparation of MLVs or ULVs depending on the choice of methods; (2) size reduction if necessary; (3) preparation of the drug solution(s) and drug loading, while this step is combined with step 1 in the case of passive drug loading; (4) buffer exchange and concentration if necessary; (5) sterile filtration or aseptic processing; (6) lyophilization, if needed, and packaging.

The potential manufacturing processes of the marketed liposomal products, summarized based on the related patents or publications.
4.1. Liposome Preparation
4.1.1. film-hydration method.
The thin-film hydration method is a traditional technique and is beneficial for loading the lipophilic drug. A thin film is created by evaporating the lipid–solvent solution during flask rotation under vacuum. MLVs suspension can be obtained by adding the aqueous solution to hydrate the lipid film. The particle size can be further reduced to obtain SUVs, and the drug substance can be passively or actively loaded during or after the liposome formation, respectively. The commercial products of AmBisome, Visudyne, and Shingrix (Adjuvant systemAS01 B ) adopt this method for manufacturing [ 56 , 77 , 80 ]. For example, Visudyne is manufactured through evaporating the ingredients from dichloromethane, hydration with lactose solution, size reduction by homogenization, filtration, and lyophilization. Adjuvant systemAS01 B is an individual vial in the product of Shingrix, and is a liposome-based adjuvant containing two immunoenhancers, QS21 (a triterpene glycoside purified from the bark of the tree Quillajasaponaria Molina) and MPL (3-Odesacyl-4′-monophosphoryl lipid A). The MPL and other lipids are dissolved in the organic solution and dried. After hydration and size reduction, the QS21 aqueous solution is added for formulation.
4.1.2. Double-Emulsification Method
This technique, also known as DepoFoamplatform TM , has been adopted by three commercial products of DepoCyte, DepoDur, and Expel to produce MVLs. The whole production routinely includes four sequential operations as follows: (1) the formation of a “water-in-oil” emulsion, (2) the formation of a “water-in-oil-in-water” emulsion, (3) solvent extraction with the help of stripping gas or vacuum pressure, and (4) microfiltration for the removal of the free drug, concentration, and exchange of external solution [ 37 , 63 ]. During the manufacturing process, aseptic assurance should be provided since MVLs owing the micro particle size cannot be produced as sterile batches through the 0.22 µm filtration. Lu et al. [ 81 ] studied the influence of the process on critical quality attributes of bupivacaine MVLs and found that the particle size of the first emulsion increases with the increase in lipid concentration, and shearing speed strongly influences the particle size. For the second emulsion, the encapsulation efficiency decreases during the solvent removal since some MVLs are collapsed and the drug leaks from the internal aqueous phase. Additionally, the high temperature promotes the mobility and rearrangement of lipid bilayers, resulting in lipid fusion and the collapse of the aqueous chambers.
4.1.3. Solvent Injection Techniques
For this kind of technique, lipid materials and lipophilic substances are dissolved in a water-miscible organic solvent, and then the organic phase is injected into a large amount of aqueous buffer, resulting in small unilamellar liposomes being spontaneously formed [ 82 ]. In other modified methods, two streams of solution are injected/infused through the Y-connector [ 83 ] and membrane contactors in a tubular (e.g., Shirasu Porous Glass membrane [ 84 ] or hollow fiber configuration [ 85 ]) and crossflow injection [ 86 , 87 ] device to improve the micromixing of the organic phase into the aqueous phase. The solvent rapidly diffuses in an aqueous medium, and interfacial turbulence leads to the formation of small and homogenous liposomes [ 88 ]. The particle size between 80 nm and 300 nm can be prepared depending on the preparation conditions [ 89 ], and the additional energy input for particle size reduction, such as sonication and extrusion, is not required. The organic solvent should be removed using evaporation, lyophilization, dialysis, or diafiltration, and the liposomes suspensions can be concentrated to the desired volume. Ethanol is commonly used as an organic solvent because of its safety. Various preparation parameters, including the flow rate, the temperature of both solvent and aqueous solution, the lipid concentration, as well as the stirring rate, can affect the properties of particles [ 88 ]. Arikayce uses “ethanol infusion” or “in-line infusion” to prepare amikacin liposomes [ 64 ]. The minimal amount of lipids–ethanol solution and the amikacin sulfate aqueous solution are mixed by a Y-connector and in-line mixer to form the nanosized amikacin liposomes.
4.1.4. In Situ Preparation of Liposomes
“In situ” is regarded as liposomes that are formed before clinical use [ 90 ]. The commercial product of Mepacthas adopted this method for production. Drug and phospholipids are formulated into a bulk solution, and filtration for sterilization, filling, and lyophilization is followed. In the case of Mepact, only three components, i.e., active ingredient muramyl tripeptide phosphatidyl ethanolamine (MTP-PE), palmitoyl-oleoyl-phosphatidylcholine (POPC) and dioleoyl-phosphatidylserine (OOPS) with a certain ratio (POPC:OOPS = 7:3, MTP-PE:phospholipids = 1:250) are included [ 91 ]. The product is a dry lipid cake with a porous structure, providing a large surface area for contact with the constitution medium. Before clinical use, 0.9% saline solution is added into the vial, and the dry substance is hydrated to form multilamellar liposomes with a particle size of 2.0–3.5 µm [ 92 ] and monomodal size distribution. The phase transition temperature of phospholipids in water is about 5 °C, which allows liposome preparation in situ at room temperature [ 93 ].
4.2. Size-Reduction Techniques
Size and size distribution are critical attributions for the performance and safety of liposomes. Several methods are available for the size reduction of liposomes, such as (ultra)sonication either by bath or probe, French press [ 94 ], extrusion, homogenization, or combination methods, such as freeze–thaw extrusion [ 95 ], freeze–thaw sonication, and a high-pressure homogenization–extrusion technique [ 96 ]. Among these techniques, extrusion and high-pressure homogenization (HPH) are the most popular used in pharmaceutical manufacturing.
The extrusion technique was first introduced in 1971 [ 97 ]. Liposomes of large sizes pass through the polycarbonate membranes (50 nm~5 µm) or asymmetric ceramic filter to become the smaller one with a fine size distribution. It is known that commercial nano-liposomal products, including Onivyde, Vyxeos, and Marqibo, use this method for production. This method is relatively simple, reproducible, and only moderate conditions are required. The potential mechanism of size reduction is that MLVs are ruptured at the entrance of the membrane pore and rearranged during the membrane passage [ 98 , 99 ]. The critical process parameters, such as the pore size of the polycarbonate membrane, the number of passage cycles, pressure, and flow rate, can influence the size and liposomal lamellarity [ 100 ]. Ong et al. [ 101 ] found that extrusion was the most efficient technique when comparing other different nanosizing techniques, including freeze–thaw sonication, (ultra)sonication, and homogenization. However, extrusion may decrease the liposome encapsulation and change the structure of asymmetric liposomes [ 102 ].
HPH is employed to produce various nano-formulations, such as liposomes, nanocrystals, and nanoemulsions. It is suitable for both aqueous and non-aqueous systems and provides different production scales, from the laboratory scale with 10 L/h capacity to large production scales with 100,000 L/h capacity [ 103 ]. Commercial liposomal products, including Visudyne and AmBisome, use this method for manufacturing. The MLVs suspension is passed through a narrow gap under high pressure, broken down by means of shear force, turbulence, and cavitation of fluid generated by the velocity gradient, and then rearranged into smaller liposomes. The particle size and size distribution are decided by the parameters of the homogenization process, such as pressure, processed cycles, valve and impingement design, and flow rate; they are also affected by the properties of samples, including the composition and viscosity of the bulk medium and initial size distribution of particles. The increasing pressure and processed cycles decrease the particle size and polydispersity index (PDI), but also resulting in a decrease in the encapsulation efficiency [ 104 , 105 ].
4.3. Drug-Loading Methods
High drug loading is desirable to minimize the amount of excipient, reach the desired concentration of therapeutic agents, decrease the dose volume, and reduce dosing time. Two primary techniques are routinely used for drug loading, i.e., passive and active drug loading procedures. Additionally, there are some other drug-entrapment methods, such as a drug–lipid chemical conjugate, the combination of passive and active drug encapsulation.
4.3.1. Passive Drug-Loading Approach
The passive drug-loading method involves encapsulating the drug agent during the preparation of liposomes. The drug can be encapsulated within the inner aqueous space or embedded in the bilayer of liposomes by means of covalent, ionic, electrostatic, non-covalent, or steric interactions between drug molecules and lipids. The main disadvantage of this approach is the low encapsulating efficiency, and thus leading to an additional step of free drug removal. From the learn of patents and publications, the marketed liposomal products using the passive drug loading method include AmBisome, Visudyne, Arikayce, DepoCyte, DepoDur, and Expel.
For Lipophilic Drug Substance
Verteporfin, also known as Benzporphyrin Derivative Monoacid Ring A (BPD) (Visudyne), is a highly lipophilic molecule, which can promote drug participation efficiently into the lipid bilayer. The entrapment efficiency of BPD in liposomes is almost 100% after homogenization [ 43 ].
AmpB (AmBisome) is poorly soluble in aqueous and in most organic solvents because of its amphipathic structure. AmpB can be tightly intercalated into the lipid bilayer by the ionic association between the positively charged amino group of AmpB and the negatively charged phosphate group of DSPG. The ionic interaction is easily formed in an acid environment of pH 1.0–3.0 [ 106 ]. In addition, the association is further strengthened by the hydrophobic interactions between the polyene portion of AmpB and aliphatic hydrocarbon chains of phospholipids.
For a Hydrophilic Drug Substance
Amikacin sulfate (Arikayce) is a freely water-soluble, anti-infective drug. Due to the limited solubility of amikacin in ethanol, amikacin transfers to a semi-soluble, coacervated state entrapped inside the core of liposomes during the liposome preparation using ethanol infusion [ 107 ]. Surprisingly, high encapsulation efficiencies (free drug 5.2% with the optimized preparation parameters) and drug-to-lipid ratio (~0.7) were obtained [ 83 ]. The encapsulated drug exhibits a low permeability from the liposome membrane because of its multi-cationic nature, rendering stable liposomes during the circulation in blood [ 108 ].
Cytarabine (DepoCyte), Morphine (DepoDur), and Bupivacaine (Exparel) aqueous solution encapsulated in the chambers of MVLs, which consists of 94% aqueous chambers and 4% lipids) [ 109 ]; therefore, a small volume of liposome suspensions contains large quantities of drugs. In order to further improve the encapsulation efficiency and sustained release, the conversion of drug compounds from monoprotic mineral acid salts into diprotic or triprotic mineral acid salts (e.g., sulfate salt or phosphoric salt) and co-encapsulating of polyalcoholic organic acids can be used [ 110 ].
4.3.2. Active Drug-Loading Approach
The active drug loading approach, also called remote drug loading, involves loading the drug agent after empty liposomes are produced. The transmembrane gradient of pH or ion concentration is the driving force to promote the drug diffuse across the membrane into the inner core of liposomes. The drug-entrapment process takes around 5 min to 30 min, and a high loading efficiency (above 90%) can be reached.
Doxil is a typical example of drug loading based on the transmembrane gradient of ammonium sulfate ( Figure 4 a). Due to the concentration of (NH 4 ) 2 SO 4 in the core of liposomes being far higher than the external medium, the neutral molecules of DOX-NH 2 with high permeability and Octanol-to-buffer partition coefficients diffuse across the lipid bilayer and enter the liposome’s inner aqueous phase. The (DOX-NH 3 ) 2 SO 4 precipitation with a fiber-like crystalline form is generated in the core of the liposome. The low solubility of (DOX-NH 3 ) 2 SO 4 minimizes the intraliposomal osmotic pressure and thus keeps the liposome integrity.

Different mechanisms of remote drug loading. ( a ) Doxil: DOX-loaded by transmembrane gradient of (NH 4 ) 2 SO 4 concentration [ 35 ]; ( b ) Myocet, Marqibo, and DaunoXome: drug loaded by transmembrane gradient of pH; ( c ) Mepact: MDP chemically conjugated to PE through a peptide spacer, then formed liposomes with other phospholipids. ( d ) Onivyde: irinote can loaded by transmembrane gradient of the concentration of sucrosofate triethylammonium salt (TEA-SOS). One molecule of SOS can bind 8 molecules of irinotecan.
For the product of Myocet, DOX is loaded before clinical use. Transmembrane pH gradient is the driving force for DOX loading ( Figure 4 b). Myocet has three vials in one package, including vial 1—doxorubicin HCl red lyophilized powder, vial 2—liposomes suspensions in 300 mM citric acid at pH 4–5, and vial 3—sodium carbonate buffer [ 46 ]. Before clinical use, empty liposomes (vial 2) are injected into the sodium carbonate buffer (vial 3) to adjust the exterliposome medium to a pH of 7–8, and then are mixed with DOX saline solution. The neutral form of DOX molecules (pKa = 8.3) in the exterliposome medium cross the liposomal bilayer and form a unique DOX-citrate complex in the vesicle interior. The DOX-citrate complex exhibits bundles of flexible fiber, attributing to DOX monomers owning a relatively flat ring stack together to form fibers [ 111 ]. The loading efficiency is above 95%. Similar to Myocet, Marqibo also has three vials in one package. The empty liposomes have the inner aqueous phase of citrate buffer (0.3 M, pH about 4.0) [ 112 ]. Before vincristine sulfate (pKa = 5.4) loading, the external pH of liposomes is increased to about pH 7.0–7.5 by adding sodium phosphate buffer at a concentration of 14.2 mg/mL.
Different from Myocet and Marqibo, DaunoXome employs a low pH gradient (citric acid, 50 mM), resulting in a relatively weaker daunorubicin loading and then a short half-life of the drug and low AUC [ 106 ]. Oppositely, a high transmembrane pH gradient (e.g., intraliposomal pH 2.0) can increase the drug encapsulation ratio and the anti-tumor efficacy of liposomes [ 113 , 114 ]. However, low pH will induce the acid-hydrolysis of lipid (such as phosphatidylcholine), further inducing the drug leakage and stability problematic of liposomes [ 115 ].
Onivyde using a novel polyanion salt, i.e., sucrosofate triethylammonium salt (TEA-SOS), to produce the electrochemical gradient across the liposomes membrane [ 116 , 117 ] ( Figure 4 d). One molecule of polyanion salt can bind eight molecules of irinotecan. The liposomes are firstly prepared in the solution of TEA-SOS. After exchanging the extraliposomal, non-encapsulated TEA-SOS medium by the drug-loading buffer, the empty liposomes are incubated with irinotecan hydrochloride solution at a pH of 6.5 [ 118 ]. Irinotecan encapsulated in the liposome interior shows a gelated or precipitated state as a sucroseoctasulfate salt form. High encapsulation efficiency of more than 95% can be obtained.
4.3.3. Drug–Lipid Conjugation by Covalently Linking
Covalently linking the drug molecules to lipids via a linker is another efficient strategy to load the drug within liposomes, e.g., Mepact [ 47 , 119 ]. Muramyl dipeptide (MDP) is the component of the cell wall of primarily Gram positive bacteria and shows the capability to enhance immune responses. The MDP liposome shows problems, including low entrapment efficiency and drug leakage during the storage since MDP is a water-soluble and low-molecular-weight molecule [ 120 ]. To improve the lipid solubility of MDP, MTP-PE (muramyl tripeptide-phosphatidyl ethanolamine) was synthesized by linking the MDP to PE through a peptide spacer [ 47 , 121 ] ( Figure 4 c). The amphipathic molecules of MTP-PE intercalated into the membrane bilayers of liposomes during the reconstitution of the lyophilized product (MTP-PE, POPC, and OOPS) with saline solution. MTP-PE existed within the liposomes, and no free MTP-PE was found [ 93 ].
4.3.4. Combination Method
A combination of passively loading and actively loading is used for Vyxeos, which is the first approved liposome loaded with two different drugs (cytarabine and daunorubicin) in the same vesicle [ 122 ]. In brief, cytarabine is passively encapsulated into liposomes when hydrating the lipids foams with a solution of Cu(gulconate) 2 , triethanolamine (TEA), pH 7.4, and cytarabine. After sizing reduction and buffer exchange to remove the unencapsulated drug and Cu(gulconate) 2 /TEA, daunorubicin buffer solution at neutral pH is incubated with the cytarabine-loaded liposomes. The daunorubicin is actively accumulated inside liposomes using a Cu(gulconate) 2 /TEA-based loading approach. The daunorubicin diffuses through the lipid bilayer into intraliposome while the neutral form of TEA permeates towards the extraliposomal medium, establishing a kinetic and stoichiometric relationship between daunorubicin and the TEA efflux [ 123 , 124 ]. The Cu(gulconate) 2 /TEA plays a key role in interacting with both drugs, keeping both drug retention inside liposomes and modulating the drug release from the liposomes [ 36 ].
5. Critical Quality Attributions
Different with the conventional drug dosage form (e.g., injection solution for small molecules), the transport of therapeutic molecules loaded in liposomes to tumor cells after systemic administration (e.g., intravenous injection) is more complex and mainly undergo the following steps [ 125 , 126 ]: (a) Extravasation from intravascular space to tissue interstitium: liposomes across the discontinuous endothelial junctions (100 nm–2 µm) of tumor vascular wall via diffusion and/or convection to enter the tumor interstitium. Meanwhile, a part of liposomes are cleared from systemic circulation by MPS, especially for the particles with a large size (>200 nm), hydrophobic and charged particle surface (negative or positive charge). (b) Interstitial transport by diffusion and convection to close to the individual tumor cells. Surface modification on liposomes using active targeting will overcome the physical resistance for particle diffusion in extracellular matrix (ECM) since higher affinity are generated between targeting ligand on the particles and the receptors on the surface of tumor cells. (3) Attach to cell membrane through non-specific or specific binding. (4) Enter the cell through the endocytosis, membrane fusion or diffusion. The pathways of endocytosis is depended on the particle size, i.e., the particles with the size of 200 nm and 500 nm are clathrin- and caveolae-mediated endocytosis, and up to 5 µm for macropinocytosis. (5) Intracellular trafficking and drug release. Based on this transporting process of liposomes, Doxil pronouncedly reduces the cardiac toxicity compared to the administration of conventional doxorubicin since the circulating liposome particles are unable to cross the continuous endothelial junctions of blood vessels in the heart [ 127 ]. DaunoXome increases daunorubicin tumor delivery by about 10-fold over conventional drug and provides sustained release in vivo [ 21 ].
Critical quality attributions (CQAs) of the product are physical, chemical, biological, or microbiological properties or characteristics that would affect the product’s pharmacokinetic and pharmacodynamic performance [ 128 ]. Based on the mechanism of pharmacokinetics and the properties of liposomes, CQAs of liposomes typically include particle size and size distribution, morphology, lamellar structure, surface properties (zeta potential, PEGlated thickness, and targeting molecules, such as a ligand, if applicable), the phase transition temperature of the lipid membrane, drug loading efficiency, release rate, etc. For example, the lamellar structure of liposomes could affect the rate of drug release, and the morphology could affect the circulation time of liposomes in vivo. Here, we focus on three CQAs in this section.
5.1. Particle Size and Size Distribution
As mentioned above, the whole pharmacokinetic process of liposomes, such as systemic circulation and the clearance by MPS, the extravasation into tissue interstitium, interstitial transport in the extracellular matrix, and cellular uptake and intracellular trafficking, are dimension-dependent [ 129 , 130 ]. The particles with a size <200 nm decrease the opsonization by serum proteins and clearance of MPS. For Myocet, smaller liposomes have higher anti-tumor efficacy and increased mean survival time in a murine leukemia model [ 46 ]. Mepact with a particle size of 2.0–3.5 µm can prompt the phagocytosis by monocytes/macrophages and trigger the immunomodulatory effects for cancer therapy [ 92 ]. Singh et al. [ 131 ] found that vaccines with different particle sizes of adjuvant liposomes (the Army Liposome Formulation, ALF) resulted in different immune responses, i.e., dendritic cells more efficiently uptake small-size particles in the range of 10–200 nm, whereas other immune cells, such as macrophages, are prone to phagocytose large-size particles. Niu et al. [ 17 ] studied insulin-loaded liposomes for oral administration and found that liposomes with the diameters of 150 nm and 400 nm exhibited slower and prolonged hypoglycemic action up to 24 h, while liposomes with a particle size of about 80 nm and 2 µm exhibited a transient and no pharmacological effect, respectively.
5.2. Surface Modification
Liposomes coated by highly flexible PEG chains to create a hydration layer are an important tool for liposome modification (described in the Section 3.2 ), which reduces the clearance by MPS, prolongs the circulation lifetime, and prevent liposomes from aggregation [ 132 ]. Another common surface modification of liposomes is using ligands for active targeting (described in the Section 1 ).
FDA guidelines recommend that the coating thickness of nanomaterial could be described in the dossier [ 133 ] since the coverage density and thickness of the layer affect cellular uptake and control nanoparticle transport through biological matrices [ 134 ]. The reflection paper of EMA [ 135 ] mentioned that the influences of the surface coating either by a non-covalent or covalently bound on the product stability, pharmacokinetics, bio-distribution, bimolecular interaction, and receptor-mediated cellular interaction should be considered. Additionally, the coating material should be completely characterized and controlled, including its consistency and reproducibility, surface coverage heterogeneity, the orientation and conformational state of the ligand, physico-chemical stability, premature detachment, and/or degradation of the coating, etc.
5.3. Phase Transition Temperature
The phase transition temperature of the bilayer membrane is a critical parameter for liposome production, stability during the storage, and drug release in vivo. A large number of investigations on the phase transition have been completed [ 65 , 136 , 137 , 138 ]. Hydrated lipid bilayers exhibit three lamellar forms: a crystal phase ( L C ), a “solid” gel phase ( L β : hexagonal lattice untitled chain or L β ’ : quasi hexagonal array with titled chain), and a liquid-crystal phase ( L α ) [ 139 ], shown in Figure 5 . In the lamellar gel phase, the acyl chains are preferentially aligned in an all-trans conformation, and lateral diffusion is very slow. Cooling under the transition temperature of Tc, the lamellar changes from a gel phase to L C phase. L C is also called the subgel phase; the hydrocarbon chains are in a fully extended, all-trans conformation, and the polar head groups are relatively immobile. Between the transition from the gel phase to L C , the metastable precursor SGII phase (also known as sub-subgel) or L R1 phase might occur [ 140 ]. Heating the temperature over the T m (melting transition temperature), the membrane changes from the order state (gel state) to a relatively disordered state ( L α ), and the hydrocarbon chains show rapid trans-gauche fluctuations, leading to an increase in the permeability of membrane and drug molecules cross the membrane easily.

The phase transition of liposomal bilayer dispersed in aqueous solution. Heating above the melting temperature (T m ), the phase of bilayer transits from “solid” gel phase ( L β : hexagonal lattice untitled chain or L β’ : quasi hexagonal array with titled chain) (ordered state) to liquid crystalline phase ( L α ) (disordered state). Cooling below T c , the phase of bilayer transits from “solid” gel phase ( L β or L β’ ) (ordered state) to subgel phase or crystalline phase ( L c ) (ordered state).
Normally, a higher T m of lamellar than physiological temperature (37 °C) is required. Thus, the rate of drug molecules crossing the gel state of the membrane remains slow. The burst release and drug leakage from liposomes in vivo can be better prevented in order to reduce the risk of systemic toxicity.
6. Regulatory Consideration
Over the last few decades, approximately 100 nanomedicines and 11 nanomedicines have been approved by the FDA and EMA, respectively, while 48 nanomedicines are presently under clinical trials in the European Union [ 109 ]. Considering the increasing number of nanomedicine applications and sharing the experience of the regulatory network in the scientific evaluation of nanomedicines, several guidelines about nanomaterial and nanoproducts were released by the FDA, EMA, Ministry of Health, Labour and Welfare—Japan (MHLW), and Chinese National Medical Products Administration (NMPA). These guidelines involve different nano-dosage forms, including liposomes, iron-based nano-colloidal products, block copolymer micelles, and nucleic-acid (siRNA)-loaded nanoproducts ( Table 3 ). Among these guidelines, all four regulatory agencies worked out the guidance about liposomes, which might be attributed to the relatively common dosage form and the relatively large numbers approved in market and clinical trials.
Nanomedicine guidelines published by FDA, EMA, MHLW, and NMPA.
Given the structure’s complexity and great diversity in liposomal products, the ultimate goal of quality, safety, and effectiveness should be kept in mind in each stage of the product’s life cycle. Based on these guidelines, we emphasize that building a comprehensive knowledgebase to better understand potential risk generated during manufacturing, analysis, and material control on the physicochemical and biological characteristics of a product is extremely important. Knowledge can be gained from the early stages of pharmaceutical research and development, can also be updated from subsequent manufacturing and associated control strategy over time. The deeper understanding of the relationships among critical material attribution (CMA), critical process parameters (CPP), physicochemical properties, and in vivo performance of liposomes, the lower risk yields [ 128 ]. Secondly, excipient, especially for lipids, plays an important role in the quality of liposomal products. A minor change of lipid materials might induce the variation of pharmacokinetic or pharmacodynamics of the drug, potentially leading to serious toxicity. The requirements of lipid control, including lipid source (extraction or synthesis), characteristic, specification, and stability, are described in detail in FDA guidance. Thirdly, sterilization is considered a challenging process for liposome manufacturing since most liposomal products are intended for parenteral administration. Sterile filtration using 0.22 µm membrane is commonly adopted in the pharmaceutical industry. However, issues such as membrane clogging, reduced integrity of liposomes, as well as ineffective retention of small bacteria may occur [ 141 ]. Therefore, the promising sterilization method and the validation of the sterilization process are critical for batch consistency as well as sterility assurance of liposome products.
In addition, the in vivo fate of liposomal carriers is another critical consideration for the development of liposome preparations because the leakage of the payload from nanoparticles may be even quicker than we have recognized [ 142 ]. A guideline for non-clinical pharmacokinetics of nanomedicine recently released by the Center for Drug Evaluation (CDE), NMPA, encourages in vivo measurement of vehicles besides cargos [ 143 ]. Fluorescent labeling is the most commonly used technique to monitor the in vivo transport of vehicles. However, it is crucial to discriminate intact vehicles from free fluorescent molecules that are released from nanoparticles [ 144 ]. Aggregation-caused quenching (ACQ)isa promising method to eliminate free-probe interference due to the environment-responsiveness characteristics, although the phenomenon was generally regarded to be unfavorable in bioimaging [ 145 ].The ACQ probes emit near-infrared fluorescence when they are loaded in carrier matrix (molecularly dispersed in general), but quench immediately and absolutely once they are released into the aqueous environment due to π-πstacking. Therefore, the fluorescence indicates the intact vehicles. The in vivo fates of various nanoparticles (e.g., polymeric nanoparticles, micelles, nanoemulsions, and nanocrystals) via different routes (e.g., oral, intravenous, transdermal, nasal, and ocular routes) have been explored by using the ACQ probes [ 146 , 147 , 148 , 149 ].
7. Future Perspectives and Concluding Remarks
We compared the number of publications setting TITLE-ABS-KEY as “liposome”, “(liposome AND medicine) or (liposome AND drug)”, “(nano AND liposomes AND medicine) or (nano AND liposomes AND drug), and “(nano AND medicine) or (nano AND drug)” in the year range between 1970 and 2020 in Scopus. Interesting results were found. From Figure 6 , we can conclude that (1) liposome as a drug carrier and applied in other fields (e.g., food, cosmetics) starts earlier than nanomedicines, i.e., the year 1970 vs. 1990. (2) The application of liposomes as medicine carriers in total liposomal publications increases over time, i.e., 50% in 2000, 70% in 2010, and 74% in 2020. (3) Although the development of nanomedicine started later than the use of liposomes, the number of publications about nanomedicine shows an exponential increase over time. (4) Extremely low percentages (7%) of medicine/drug nanoliposomes in total nanomedicine/drug are observed, which might be false data, since there are 3024 publications about liposome medicine in the year 2020. We speculate that the name combination of “nano” and “liposome” might be less frequently used compared with the names of nanoparticles, nanocrystals, or nanosuspensions. It was reported by the FDA that more than 500 liposome applications were received up until 18 February 2016 [ 79 ]. Among these applications, except for around 100 submissions applied for combination therapy of liposome with another therapeutic, the remaining submissions (used individually) were 3% NDAs, 1% ANDAs, and 96% INDs. The data collected from the laboratory level and pharmaceutical industry indicate that there will be a large number of liposomal products transformed from the laboratory bench to pilot plant and market in the near future.

The comparison profiles of publications using setting TITLE-ABS-KEY as “liposome”, “(liposome AND medicine) or (liposome AND drug)”, “(nano AND liposomes AND medicine) or (nano AND liposomes AND drug) and “(nano AND medicine) or (nano AND drug)” in the year range between 1970 and 2020 in Scopus.
From the first liposome product of Doxil approved in 1995, liposome techniques have been further developed for more than 20 years. We summarize the successful experience and pain points based on the abundance of publications and commercial products. Liposomes can be well-designed and display intended functions depending on human requirements and needs. On the one hand, there are major obstacles during the development and commercialized production, such as the individual differences in the EPR effect, accelerated blood clearance (ABC) phenomenon of PEGylated liposomes, scale-up, the reproducibility/consistency among different batches and manufacturing sites, and excipient control.
On the other hand, a large number of smart liposomal systems are developing in laboratory or undergoing clinical trials, such as active targeting liposomes (e.g., anti-EGFR immunoliposomes, phase II; MBP-426, phase II) and stimuli-sensitive liposomes (e.g., ThermoDox) [ 150 ]. The microenvironment at the target disease site can be exploited to trigger the release of drug from liposome carrier. The external or internal stimuli such as temperature, pH, light, eletromagnetic fields, enzyme, and hypoxia, are frequently studied as “on-off” switch of the drug release [ 151 , 152 ]. Although promising results were obtained in pre-clinical studies, it is challenging for successful clinical translation due to the major issues, such as the leakage of the cargo before reaching the target sites, the individual differences among patients, as well as the multi-modal therapies involved. ThermoDox, the fastest developed thermosensitive liposomes, got a failure at the second Phase III clinical trial, designed for combination with radiofrequency ablation for the treatment of hepatocellular carcinoma [ 153 ]. However, the failure just means the failure of liposomal product under the certain clinical design, and these smart techniques will present many new opportunities for liposomes to further increase the therapeutic efficacy and decrease the side-effects.
A topic about “where are we in the development path of nanomedicines” has been widely argued recently. When looking back through history on the application situation and the performance of liposomes, we maintain a positive attitude. Three types of liposomal products have been approved by the Chinese NMPA, i.e., Lipusu (paclitaxel liposome), doxorubicin hydrochloride liposome, and Amphotericin Bliposome. Additionally, both large pharmaceutical industries and small innovative companies in China are developing nanomedicines, including liposomes, nanocrystal, inorganic particles, and polymeric micelles. At the same time, the topic of the “safety and quality evaluation of nanomedicine” has been selected as a key project for developing the regulatory science by NMPA in 2019. We are preparing a regulatory framework to respond to the future of nanomedicines with a seamless connection.
Acknowledgments
We are grateful to Min Huang, Shengyang Pharmaceutical University, for supporting us in preparing the figures.
Author Contributions
Writing—original draft preparation, P.L.; Review and editing, J.Z.; Supervision, G.C. All authors have read and agreed to the published version of the manuscript.
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Conflicts of interest.
The authors declare no conflict of interest.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Liposomes: structure, composition, types, and clinical applications
Affiliations.
- 1 Pharmacological and Diagnostic Research Center, Faculty of Pharmacy, Al-Ahliyya Amman University, Amman, 19328, Jordan.
- 2 Department of Chemistry, Faculty of Arts and Science, Applied Science Private University, Amman, Jordan.
- 3 Department of Biology, The University of Jordan, Amman, 11942, Jordan.
- 4 Department of Chemistry, The University of Jordan, Amman, 11942, Jordan.
- 5 Hamdi Mango Center for Scientific Research, The University of Jordan, Amman, 11942, Jordan.
- 6 Cell Therapy Center, The University of Jordan, Amman, 11942, Jordan.
- PMID: 35600452
- PMCID: PMC9118483
- DOI: 10.1016/j.heliyon.2022.e09394
Liposomes are now considered the most commonly used nanocarriers for various potentially active hydrophobic and hydrophilic molecules due to their high biocompatibility, biodegradability, and low immunogenicity. Liposomes also proved to enhance drug solubility and controlled distribution, as well as their capacity for surface modifications for targeted, prolonged, and sustained release. Based on the composition, liposomes can be considered to have evolved from conventional, long-circulating, targeted, and immune-liposomes to stimuli-responsive and actively targeted liposomes. Many liposomal-based drug delivery systems are currently clinically approved to treat several diseases, such as cancer, fungal and viral infections; more liposomes have reached advanced phases in clinical trials. This review describes liposomes structure, composition, preparation methods, and clinical applications.
Keywords: Lamellarity; Liposomes; Phospholipids; Stealth liposomes; Vaccinations.
© 2022 The Author(s).
Publication types
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- News & Views
- Published: 19 September 2012
Materials chemistry
Liposomes derived from molecular vases
- Cyrus R. Safinya 1 &
- Kai K. Ewert 1
Nature volume 489 , pages 372–374 ( 2012 ) Cite this article
7531 Accesses
60 Citations
50 Altmetric
Metrics details
- Drug delivery
Liposomes are ubiquitous components of skin moisturizers and other personal-care products. Modified liposomes prepared from receptor-like molecules open up fresh opportunities for therapeutic and industrial applications.
This is a preview of subscription content, access via your institution
Relevant articles
Open Access articles citing this article.
Recent advances in ocular lubrication
- Jianhua Zhang
- , Yunjuan Su
- … Hongdong Wang
Friction Open Access 08 February 2024
Nanomaterials for application in wound Healing: current state-of-the-art and future perspectives
- Manal Aly Shalaby
- , Mohammed Moustapha Anwar
- & Hesham Saeed
Journal of Polymer Research Open Access 19 February 2022
Paclitaxel loading in cationic liposome vectors is enhanced by replacement of oleoyl with linoleoyl tails with distinct lipid shapes
- Yuhong Zhen
- , Kai K. Ewert
- … Cyrus R. Safinya
Scientific Reports Open Access 31 March 2021
Access options
Subscribe to this journal
Receive 51 print issues and online access
185,98 € per year
only 3,65 € per issue
Rent or buy this article
Prices vary by article type
Prices may be subject to local taxes which are calculated during checkout
Kubitschke, J., Javor, S. & Rebek, J. Chem. Commun. 48 , 9251–9253 (2012).
Article CAS Google Scholar
Cram, D. J. Science 219 , 1177–1183 (1983).
Article CAS ADS Google Scholar
Bangham, A. D. & Horne, R. W. J. Mol.Biol. 8 , 660–668 (1964).
Lasic, D. D. Liposomes: From Physics to Applications (Elsevier, 1993).
Google Scholar
Papahadjopoulos, D. in Stealth Liposomes (eds Lasic, D. D. & Martin, F.) 1–6 (CRC, 1995).
Felgner, P. L. et al. Proc. Natl Acad. Sci. USA 84 , 7413–7417 (1987).
Ewert, K. K. et al. Top. Curr. Chem. 296 , 191–226 (2010).
Nabel, G. J. et al. Proc. Natl Acad. Sci. USA 90 , 11307–11311 (1993).
www.wiley.com/legacy/wileychi/genmed/clinical
Huang, L., Hung, M.-C. & Wagner, E. (eds) Non-Viral Vectors for Gene Therapy 2nd edn, Part I (Academic, 2005).
Safinya, C. R., Ewert, K. K. & Leal, C. Liq. Cryst. 38 , 1715–1723 (2011).
Rädler, J. O., Koltover, I., Salditt, T. & Safinya, C. R. Science 275 , 810–814 (1997).
Article Google Scholar
Liu, Y., Liao, P., Cheng, Q. & Hooley, R. J. J. Am. Chem. Soc. 132 , 10383–10390 (2010).
Feher, K. M., Hoang, H. & Schramm, M. P. N. J. Chem. 36 , 874–876 (2012).
Discher, B. M. et al. Science 284 , 1143–1146 (1999).
Download references
Author information
Authors and affiliations.
Cyrus R. Safinya and Kai K. Ewert are in the Departments of Materials, Physics, and Molecular, Cellular and Developmental Biology, University of California, Santa Barbara, Santa Barbara, California 93106, USA. They are also affiliated with the Materials Research Laboratory, University of California, Santa Barbara.,
Cyrus R. Safinya & Kai K. Ewert
You can also search for this author in PubMed Google Scholar
Corresponding authors
Correspondence to Cyrus R. Safinya or Kai K. Ewert .
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Safinya, C., Ewert, K. Liposomes derived from molecular vases. Nature 489 , 372–374 (2012). https://doi.org/10.1038/489372b
Download citation
Published : 19 September 2012
Issue Date : 20 September 2012
DOI : https://doi.org/10.1038/489372b
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
- Hongdong Wang
Friction (2024)
Nanomedicine-based treatment: An emerging therapeutical strategy for pulmonary hypertension
- Qiaohui Chen
- Changping Hu
Nano Research (2023)
- Mohammed Moustapha Anwar
- Hesham Saeed
Journal of Polymer Research (2022)
Enhancement strategies for transdermal drug delivery systems: current trends and applications
- Delly Ramadon
- Maeliosa T. C. McCrudden
- Ryan F. Donnelly
Drug Delivery and Translational Research (2022)
- Kai K. Ewert
- Cyrus R. Safinya
Scientific Reports (2021)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Translational Research newsletter — top stories in biotechnology, drug discovery and pharma.

Advertisement
QbD-assisted optimisation of liposomes in chitosan gel for dermal delivery of aceclofenac as synergistic approach to combat pain and inflammation
- Original Article
- Published: 30 January 2024
Cite this article
- Ghanshyam Das Gupta 1 ,
- Harmanpreet Singh 2 ,
- Shamsher Singh 3 &
- Amrinder Singh ORCID: orcid.org/0000-0002-1724-1420 1 , 4
131 Accesses
Explore all metrics
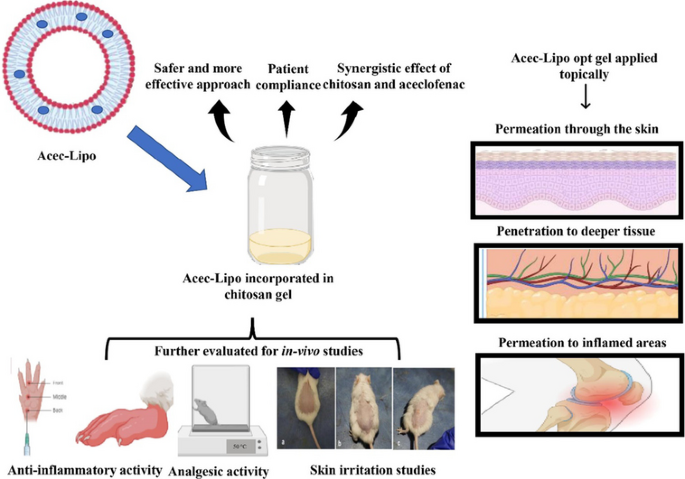
Aceclofenac (ACE) is a drug that was precisely devised to circumvent the shortcomings associated with diclofenac. However, ACE too corresponds to nonsteroidal anti-inflammatory drug (NSAID)-related adverse effects, but with a lower amplitude. The present investigation seeks to develop liposomes loaded with ACE adopting a central composite design (CCD) and formulate a chitosan-based hydrogel for synergistic anti-inflammatory efficacy and improved ACE dermal administration. On the basis of preliminary vesicle size, Poly Dispersity Index (PDI), and drug entrapment, the composition of lipid, cholesterol, and vitamin E TPGS were chosen as independent variables. The formulation composition met the specifications for an optimum liposomal formulation, with total lipid concentration (13.5% w/w), cholesterol concentration (10% w/w), and surfactant concentration (2% w/w). With particle size and PDI of 174.22 ± 5.46 nm and 0.285 ± 0.01 respectively, the optimised formulation achieved an entrapment effectiveness of 92.08 ± 3.56%. Based on the CCD design, the optimised formulation Acec-Lipo opt was chosen and was subsequently transformed to a chitosan-based gel formulation for in vitro drug release, penetration through the skin, in vivo analgesic therapeutic activity, and skin irritation testing. % age oedema inhibition was found to be greatest with the Acec-Lipo opt gel formulation, followed by Acec gel. These results reinforce the notion that the inclusion of chitosan resulted in a synergistic effect despite the same strength of the drug. The findings suggested that Acec-Lipo incorporated in chitosan gel for skin targeting might be an effective formulation for topical ACE administration in clinical subjects.
Graphical abstract
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
Similar content being viewed by others

Application of 32 factorial design for loratadine-loaded nanosponge in topical gel formulation: comprehensive in-vitro and ex vivo evaluations
Durgaramani Sivadasan, Krishnaraju Venkatesan, … Sami El Deeb

Optimization and Characterization of Thymoquinone-Loaded Liposomes with Enhanced Topical Anti-inflammatory Activity
Mahmoud Mostafa, Eman Alaaeldin, … Hatem A. Sarhan
Formulation Optimization and Ex Vivo and In Vivo Evaluation of Celecoxib Microemulsion-Based Gel for Transdermal Delivery
Mengyuan Cao, Lili Ren & Guoguang Chen
Availability of data and materials
The data is available with the first author and corresponding author and the materials were provided by the institute.
Raj R, Mongia P, Ram A, Jain NK. Enhanced skin delivery of aceclofenac via hydrogel-based solid lipid nanoparticles. Artif Cells Nanomed Biotechnol. 2016;44:1434–9.
Article CAS PubMed Google Scholar
Iolascon G, Gimenez S, Mogyorosi D. A review of aceclofenac: analgesic and anti-inflammatory effects on musculoskeletal disorders. J Pain Res. 2021;14:3651–63.
Bindu S, Mazumder S, Bandyopadhyay U. Non-steroidal anti-inflammatory drugs (NSAIDs) and organ damage: a current perspective. Biochem Pharmacol. 2020;180: 114147.
Article CAS PubMed PubMed Central Google Scholar
Jeong WY, Kwon M, Choi HE, Kim KS. Recent advances in transdermal drug delivery systems: a review. Biomater Res. 2021;25:24.
Article PubMed Google Scholar
Amisha, Singh D, Kurmi BD & Singh A (2023). Recent advances in nanocarrier-based approaches to atopic dermatitis and emerging trends in drug development and design. Curr Drug Deliv .
Yang C, Wu T, Qi Y, Zhang Z. Recent advances in the application of vitamin E TPGS for drug delivery. Theranostics. 2018;8:464–85.
Fong D & Hoemann CD (2018). Chitosan immunomodulatory properties: perspectives on the impact of structural properties and dosage. Future Sci OA, 4 : FSO225.
Garg NK, Sharma G, Singh B, Nirbhavane P, Tyagi RK, Shukla R, Katare OP. Quality by Design (QbD)-enabled development of aceclofenac loaded-nano structured lipid carriers (NLCs): an improved dermatokinetic profile for inflammatory disorder (s). Int J Pharm. 2017;517(1–2):413–31.
Alam P, Shakeel F, Foudah AI, Alshehri S, Salfi R, Alqarni MH & Aljarba TM (2022). Central composite design (CCD) for the Optimisation of Ethosomal Gel Formulation of Punica granatum extract: in vitro and in vivo evaluations. Gels, 8.
Jain P, Taleuzzaman M, Kala C, Kumar Gupta D, Ali A, Aslam M. Quality by design (Qbd) assisted development of phytosomal gel of aloe vera extract for topical delivery. J Liposome Res. 2021;31:381–8.
Xu X, Khan MA, Burgess DJ. A quality by design (QbD) case study on liposomes containing hydrophilic API: II. Screening of critical variables, and establishment of design space at laboratory scale. Int J Pharm. 2012;423:543–53.
Singh A, Vaish A, Shukla R. Box-Behnken design optimized silibinin loaded glycerylmonooleate nanoliquid crystal for brain targeting. Chem Phys Lipid. 2022;1(244): 105193.
Article Google Scholar
Chopra H, Dey PS, Das D, Bhattacharya T, Shah M, Mubin S, Maishu SP, Akter R, Rahman MH, Karthika C, Murad W. Curcumin nanoparticles as promising therapeutic agents for drug targets. Molecules. 2021;26(16):4998.
Singh A, Thakur S, Singh H, Singh H, Kaur S, Kaur S, Dudi R, Mondhe DM, Jain SK. Novel vitamin E TPGS based docetaxel nanovesicle formulation for its safe and effective parenteral delivery: toxicological, pharmacokinetic and pharmacodynamic evaluation. J Liposome Res. 2021;31:365–80.
Jiang C, Ma R, Jiang X, Fang R & Ye J (2023). A transfersomes hydrogel patch for cutaneous delivery of propranolol hydrochloride: formulation, in vitro, ex vivo and in vivo studies. J Liposome Res : 1–10.
Hashemi SH, Montazer M, Naghdi N, Toliyat T. Formulation and characterization of alprazolam-loaded nanoliposomes: screening of process variables and optimizing characteristics using RSM. Drug Dev Ind Pharm. 2018;44:296–305.
Ge Y, Ge M. Development of tea tree oil-loaded liposomal formulation using response surface methodology. J Liposome Res. 2015;25:222–31.
Ali A, Ali S, Aqil M, Imam SS, Ahad A, Qadir A. Thymoquinone loaded dermal lipid nano particles: box Behnken design optimization to preclinical psoriasis assessment. J Drug Deliv Sci Technol. 2019;52:713–21.
Article CAS Google Scholar
Jangde R, Singh D. Preparation and optimization of quercetin-loaded liposomes for wound healing, using response surface methodology. Artif Cells Nanomed Biotechnol. 2016;44:635–41.
Kaur N, Kaur M, Mahajan M, Jain SK. Development, characterization and evaluation of nanocarrier based formulations of antipsoriatic drug “acitretin” for skin targeting. J Drug Deliv Sci Technol. 2020;60: 102010.
Avachat AM, Takudage PJ. Design and characterization of multifaceted lyophilized liposomal wafers with promising wound healing potential. J Liposome Res. 2018;28:193–208.
Mostafa M, Alaaeldin E, Aly UF, Sarhan HA. Optimization and characterization of thymoquinone-loaded liposomes with enhanced topical anti-inflammatory activity. AAPS PharmSciTech. 2018;19:3490–500.
Jain SK, Panchal N, Singh A, Thakur S, Shahtaghi NR, Sharma S, Guleria A. Novel self-micro emulsifying drug delivery system for safe intramuscular delivery with improved pharmacodynamics and pharmacokinetics. Curr Drug Deliv. 2021;18:1533–49.
Bachhav YG, Patravale VB. Microemulsion based vaginal gel of fluconazole: formulation, in vitro and in vivo evaluation. Int J Pharm. 2009;365:175–9.
Rai VK, Roy A, Sharma A, Rath G, Kar B, Ghosh G, Pradhan D & Halder JJJOPI (2023). Development and pharmaceutical evaluation of azelaic acid and vitamin E oil-based nanoemulgel of tacrolimus for topical application in plaque psoriasis.1–10.
Krishnaiah YS, Xu X, Rahman Z, Yang Y, Katragadda U, Lionberger R, Peters JR, Uhl K, Khan MA. Development of performance matrix for generic product equivalence of acyclovir topical creams. Int J Pharm. 2014;475:110–22.
Kulkarni M, Potdar S, Date AA, Marfatiya A. In vitro release testing of acyclovir topical formulations using immersion cells. Assay Drug Dev Technol. 2021;19:75–84.
Sharma G, Goyal H, Thakur K, Raza K, Katare OP. Novel elastic membrane vesicles (EMVs) and ethosomes-mediated effective topical delivery of aceclofenac: a new therapeutic approach for pain and inflammation. Drug Deliv. 2016;23:3135–45.
Barbosa AGR, Tintino C, Pessoa RT, De Lacerda Neto LJ, Martins A, De Oliveira MRC, Coutinho HDM, Cruz-Martins N, Quintans Junior LJ, Wilairatana P, De Menezes IRA. Anti-inflammatory and antinociceptive effect of Hyptis martiusii BENTH leaves essential oil. Biotechnol Rep (Amst). 2022;35: e00756.
Banerjee S, Chattopadhyay P, Ghosh A, Pathak MP, Singh S, Veer V. Acute dermal irritation, sensitization, and acute toxicity studies of a transdermal patch for prophylaxis against (±) anatoxin-A poisoning. Int J Toxicol. 2013;32(4):308–13.
Oecdilibrary. OECD Guidelines for the Testing of Chemicals, Section 4 [Online]. Available at: https://www.oecd-ilibrary.org/environment/test-no-402-acute-dermal toxicity_9789264070585-en.
Karal MA, Mokta NA, Levadny V, Belaya M, Ahmed M, Ahamed MK, Ahammed S. Effects of cholesterol on the size distribution and bending modulus of lipid vesicles. PLoS ONE. 2022;17(1): e0263119.
Wu Y, Xu Y, Sun WJ. Preparation and particle size controlling of papain nanoliposomes. J Shanghai Jiaotong Univ Agric Sci. 2007;25:105–9.
CAS Google Scholar
Taghizadeh SM, Bajgholi SJJOB & Nanobiotechnology (2011). A new liposomal-drug-in-adhesive patch for transdermal delivery of sodium diclofenac. 2 : 576.
Shaker S, Gardouh AR, Ghorab MM. Factors affecting liposomes particle size prepared by ethanol injection method. Research in pharmaceutical sciences. 2017;12(5):346.
Maritim S, Boulas P, Lin Y. Comprehensive analysis of liposome formulation parameters and their influence on encapsulation, stability and drug release in glibenclamide liposomes. Int J Pharm. 2021;5(592): 120051.
Torres-Flores G, Gonzalez-Horta A, Vega-Cantu YI, Rodriguez C, Rodriguez-Garcia A. Preparation and characterization of liposomal everolimus by thin-film hydration technique. Adv Polym Technol. 2020;10(2020):1–9.
Zafar A, Alruwaili NK, Imam SS, Yasir M, Alsaidan OA, Alquraini A, Rawaf A, Alsuwayt B, Anwer MK, Alshehri S & Ghoneim MM (2022). Development and optimization of nanolipid-based formulation of diclofenac sodium: in vitro characterization and preclinical evaluation. Pharmaceutics, 14.
Sankhyan A, Pawar PK. Metformin loaded non-ionic surfactant vesicles: optimization of formulation, effect of process variables and characterization. DARU J Pharma Sci. 2013;21:1–8.
Google Scholar
Kaur M, Singh K, Jain SK. Luliconazole vesicular based gel formulations for its enhanced topical delivery. J Liposome Res. 2020;30:388–406.
Raza K, Kumar M, Kumar P, Malik R, Sharma G, Kaur M, Katare OP. Topical delivery of aceclofenac: challenges and promises of novel drug delivery systems. Biomed Res Int. 2014;2014: 406731.
Article PubMed PubMed Central Google Scholar
Download references
Acknowledgements
The authors are thankful to the ISFAL and Pharmacology Department at ISF College of Pharmacy, Moga, India, for providing access to their sophisticated analytical instruments. We are grateful to DST-FIST, New Delhi, for providing infrastructure in college. We are also grateful to Punjab University Chandigarh, India, for providing HRTEM facility to perform morphological examination of the vesicular system.
Author information
Authors and affiliations.
Department of Pharmaceutics, ISF College of Pharmacy, Moga, 142 001, India
Amisha, Ghanshyam Das Gupta & Amrinder Singh
GEM Lab, Department of Pathology, Augusta University, Augusta, GA, USA
Harmanpreet Singh
Department of Pharmacology, ISF College of Pharmacy, Moga, 142001, India
Shamsher Singh
Chitkara College of Pharmacy, Chitkara University, Rajpura, Punjab, India
Amrinder Singh
You can also search for this author in PubMed Google Scholar
Contributions
AK—investigation, software, validation, formal analysis, writing—original draft. GDG—methodology, resources. HS—validation, investigation. SS—resources. AS—conceptualization, methodology, resources, writing—review and editing, supervision.
Corresponding author
Correspondence to Amrinder Singh .
Ethics declarations
Ethics approval and consent to participate.
All the ethics were adopted while conducting the research.
Consent for publication
All the authors provided their consent to publish the work.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
Reprints and permissions
About this article
Amisha, Das Gupta, G., Singh, H. et al. QbD-assisted optimisation of liposomes in chitosan gel for dermal delivery of aceclofenac as synergistic approach to combat pain and inflammation. Drug Deliv. and Transl. Res. (2024). https://doi.org/10.1007/s13346-024-01514-z
Download citation
Accepted : 02 January 2024
Published : 30 January 2024
DOI : https://doi.org/10.1007/s13346-024-01514-z
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Aceclofenac
- Topical delivery
- Skin irritation
- Skin permeation
- Anti-inflammatory activity
- Find a journal
- Publish with us
- Track your research
- Open access
- Published: 09 April 2024
Targeting transitioning lung monocytes/macrophages as treatment strategies in lung disease related to environmental exposures
- Aaron D. Schwab 1 ,
- Todd A. Wyatt 2 , 3 , 4 ,
- Grace Moravec 1 ,
- Geoffrey M. Thiele 2 , 5 ,
- Amy J. Nelson 1 ,
- Angela Gleason 1 ,
- Oliver Schanze 1 ,
- Michael J. Duryee 2 , 5 ,
- Debra J. Romberger 2 , 3 ,
- Ted R. Mikuls 2 , 5 &
- Jill A. Poole 1
Respiratory Research volume 25 , Article number: 157 ( 2024 ) Cite this article
21 Accesses
Metrics details
Environmental/occupational exposures cause significant lung diseases. Agricultural organic dust extracts (ODE) and bacterial component lipopolysaccharide (LPS) induce recruited, transitioning murine lung monocytes/macrophages, yet their cellular role remains unclear.
CCR2 RFP + mice were intratracheally instilled with high concentration ODE (25%), LPS (10 μg), or gram-positive peptidoglycan (PGN, 100 μg) for monocyte/macrophage cell-trafficking studies. CCR2 knockout (KO) mice and administration of intravenous clodronate liposomes strategies were employed to reduce circulating monocytes available for lung recruitment following LPS exposure. Lung tissues and bronchoalveolar lavage fluid (BALF) were collected. Pro-inflammatory and/or pro-fibrotic cytokines, chemokines, and lung extracellular matrix mediators were quantitated by ELISA. Infiltrating lung cells including monocyte/macrophage subpopulations, neutrophils, and lymphocytes were characterized by flow cytometry. Lung histopathology, collagen content, vimentin, and post-translational protein citrullination and malondialdehyde acetaldehyde (MAA) modification were quantitated. Parametric statistical tests (one-way ANOVA, Tukey’smultiple comparison) and nonparametric statistical (Kruskal–Wallis, Dunn’s multiple comparison) tests were used following Shapiro–Wilk testing for normality.
Intratracheal instillation of ODE, LPS, or PGN robustly induced the recruitment of inflammatory CCR2 + CD11c int CD11b hi monocytes/macrophages and both CCR2 + and CCR2 − CD11c − CD11b hi monocytes at 48 h. There were also increases in CCR2 + CD4 + and CD8 + T cells and NK cells. Despite reductions in LPS-induced lung infiltrating CD11c int CD11b hi cells (54% reduction), CCR2 knockout (KO) mice were not protected against LPS-induced inflammatory and pro-fibrotic consequences. Instead, compensatory increases in lung neutrophils and CCL2 and CCL7 release occurred. In contrast, the depletion of circulating monocytes through the administration of intravenous clodronate (vs. vehicle) liposomes 24 h prior to LPS exposure reduced LPS-induced infiltrating CD11c int CD11b hi monocyte-macrophage subpopulation by 59% without compensatory changes in other cell populations. Clodronate liposome pre-treatment significantly reduced LPS-induced IL-6 (66% reduction), matrix metalloproteinases (MMP)-3 (36%), MMP-8 (57%), tissue inhibitor of metalloproteinases (61%), fibronectin (38%), collagen content (22%), and vimentin (40%). LPS-induced lung protein citrullination and MAA modification, post-translational modifications implicated in lung disease, were reduced (39% and 48%) with clodronate vs. vehicle liposome.
Highly concentrated environmental/occupational exposures induced the recruitment of CCR2 + and CCR2 − transitioning monocyte-macrophage and monocyte subpopulations and targeting peripheral monocytes may reduce the adverse lung consequences resulting from exposures to LPS-enriched inhalants.
Introduction
Environmental and occupational lung diseases remain a significant cause of pulmonary impairment worldwide [ 1 ]. Chronic respiratory diseases including chronic obstructive pulmonary disease (COPD), asthma, asthma-like syndrome, byssinosis, hypersensitivity pneumonitis, and pulmonary fibrosis have been associated with exposure to organic dusts. Organic dusts are comprised of particulate matter, components of bacteria, fungi, viruses, pollen, and fragments of animals and plants [ 2 ]. These exposures are common to agriculture and farming, the grain and food processing industry, waste and recycling facilities, the textile and cotton industry, woodworking, concentrated urban areas, flood- and water-damaged buildings, and more [ 3 , 4 , 5 , 6 , 7 ]. Retrospective analysis of the Global Burden of Disease database identified roughly 519,100 deaths and 13.6 million disability-adjusted life years in 2016 from chronic respiratory disease due to occupational airborne exposures [ 8 ]. The incidence and prevalence of interstitial lung disease increased from 1990 to 2019, with occupational exposure implicated as a leading risk factor despite increased awareness and implementation of preventative measures [ 9 ]. However, there remains a paucity of therapeutic options aimed at hastening recovery and/or preventing chronic disease resulting from these exposures.
Lipopolysaccharide (LPS) or endotoxin is found in the outer membrane of gram-negative bacteria [ 10 , 11 , 12 ] and acts as a well-defined component of many organic dusts and disease-causing environmental exposures. A role for gram-positive cell wall components such as peptidoglycans (PGN) in mediating lung disease following organic dust exposures has also been identified. Growing industrialization, intensified agricultural production, climate change, and the rising frequency and severity of extreme weather events have conspired to increase concentrations of aerosolized organic particulate matter and environmental endotoxins [ 13 , 14 , 15 , 16 ]. Organic dusts and its bacterial components engage innate immune signaling pathways (i.e., Toll-like receptors) to initiate airway inflammatory responses marked by the influx of neutrophils, lymphocytes, monocytes, and macrophages with a corresponding release of pro-inflammatory/fibrotic mediators (e.g., tumor necrosis factor (TNF)-α, interleukin (IL)-6, chemoattractants, extracellular matrix proteins). Organic dusts and LPS exposures also induce post-translational modifications in proteins, which may serve to increase inflammation and promote tissue fibrosis [ 17 , 18 , 19 , 20 ]. Although dust and bacterial component-induced lymphocytic lung aggregates were reduced in T and B cell depleted mice, many inflammatory consequences persisted. Thus, there is growing interest in the immunopathogenic role of distinct lung monocyte-macrophage subpopulations recruited and induced following environmental exposures [ 12 , 21 , 22 ]. CC motif chemokine receptor 2 (CCR2) is a critical facilitator of monocyte recruitment and activation via interaction with its high-affinity ligand CCL2 [ 23 , 24 ]. CCR2 + monocytes play a critical role in the onset of inflammation and, upon reaching the inflamed tissue, differentiate into phenotypically and functionally distinct macrophages, capable of modulating inflammatory responses [ 25 ].
The objectives of this study were to first delineate the magnitude and distribution of CCR2 + (and CCR2 − ) monocytes and monocyte-derived macrophages trafficked to the lung following organic dust, LPS, and PGN exposures, then determine whether targeting these lung monocytes-macrophages would attenuate resulting pro-inflammatory and pro-fibrotic responses. We tested the hypotheses that pro-inflammatory and pro-fibrotic responses in lungs from mice exposed to inhaled LPS would be reduced in CCR2 deficient (knockout) mice compared to wild-type mice and that similar protection would be afforded in wild-type mice through the administration of intravenous clodronate liposome to deplete circulating monocytes. Understanding the role of recruited monocytes and monocyte-derived macrophages will provide fundamental knowledge of inflammatory processes following environmental exposures, but also potentially elucidate therapeutic targets to mitigate disease development in at-risk persons.
Environmental exposures
Lipopolysaccharide (LPS) from gram-negative Escherichia coli (O55:B5; Sigma, St. Louis, MO) served as the primary exposure in all experiments. The rationale was that LPS is commercially available and elicits a dose-dependent, reproducible pro-inflammatory lung response in humans and rodents. In studies of monocyte trafficking, comparisons were undertaken using peptidoglycan (PGN) from gram-positive Staphylococcus aureus (Sigma) and an aqueous solution of organic dust extract (ODE) prepared from swine confinement feeding facilities as previously described [ 26 ]. Briefly, settled surface dust (1 g) was incubated in sterile Hank’s Balanced Salt Solution (10 mL; Mediatech, Manassas, VA) for 1 h and centrifuged for 30 min at 2850 × g twice, with the final supernate filter-sterilized (0.22 um) to remove microorganisms and coarse particles. Stock ODE was batch prepared and stored at − 20 °C; aliquots were diluted for each experiment to a final concentration (vol/vol) of 25% in sterile phosphate buffered saline (PBS, pH 7.4; diluent). Endotoxin concentrations were determined using the limulus amebocyte lysate assay (Lonza, Walkersville, MD). Endotoxin levels averaged 1.308–2.616 μg (~ 10–50 EU) for 25% ODE. Prior mass spectrometry studies of ODE have revealed significant amounts of muramic acid (peptidoglycan marker) and 3-hydroxy fatty acids (endotoxin marker), but not ergosterol (fungi marker) as compared to house dust [ 26 ].
Animal exposure model
C57BL/6 and homozygous CCR2 RFP/RFP (B6.129(Cg)-Ccr2 tm2.1lfc /J) mice between 6 and 8 weeks of age were purchased from The Jackson Laboratory (Bar Harbor, ME). In this latter strain (#017586; RRID:IMSR_JAX:017586), a monomeric red fluorescent protein (RFP) sequence replaces the coding sequence of the Ccr2 gene, abolishing gene function and thus referred to as CCR2 knockout (KO) mice. Mouse tail snips were collected and shipped for DNA extraction and targeted CCR2 genotyping (TransnetYX, Cordova, TN) to confirm CCR2 KO. To generate heterozygous CCR2 +/RFP mice in which CCR2 is functional yet marked by RFP expression, CCR2 RFP/RFP mice were bred to C57BL/6 wild-type (WT) mice. For experiments using heterozygous CCR2 +/RFP animals, male and female mice were utilized. For the CCR2 KO (and clodronate liposome studies), male mice were utilized, as male mice had increased inflammatory responses with less experimental variability following LPS exposure, consistent with prior studies [ 12 , 27 ]. Mice were randomized, with AJN, AG, and animal facility staff aware of the randomization, whereas all other authors were blinded. To induce airway inflammation, mice were lightly sedated under isoflurane (VetOne, Boise, ID) and received one treatment with 50 μl of sterile saline (control), ODE (25%), LPS (10 μg), or PGN (100 μg) [ 28 ]. Animals were euthanized 48 h following exposure by isoflurane followed by exsanguination (right axillary blood collection). No respiratory distress, signs of stress, or significant weight loss (defined as > 20%) were observed throughout the exposure period.
Clodronate-induced systemic monocyte/macrophage depletion
In separate studies, C57BL/6 WT mice were administered encapsulated clodronate liposomes intravenously to deplete systemic monocytes and recruited monocyte-derived macrophages [ 29 , 30 , 31 , 32 ]. Clodronate and control liposomes (Liposoma Technology, Amsterdam, Netherlands; 200 μl × 5 mg/ml) were injected into the tail vein one day prior to LPS and saline control exposure.
Lavage fluid cells and lung homogenates
Bronchoalveolar lavage fluid (BALF) was collected using 3 × 1 mL PBS. Total cell numbers from the combined recovered lavage were enumerated using a BioRad TC 20 cell counter with differential cell counts determined from cytospin-prepared slides (cytopro cytocentrifuge, ELITech Group, Logan, UT) stained with Diff-Quick (Siemens, Newark, DE). Lung tissue homogenates were prepared by homogenizing lung samples (1/2 of right lungs) in 500 μl of sterile phosphate buffered saline (PBS) following removal of BALF and blood from the pulmonary vasculature. From cell-free lung tissue homogenates, levels of TNF-α, IL-6, murine neutrophil chemoattractant CXCL1, murine monocyte (and leukocyte) chemoattractants CCL2 and CCL7, and transforming growth factor (TGF)-β were quantitated by ELISA (R&D Systems, Minneapolis, MN) with minimal detectable difference (MDD) of 1.88, 1.6, 2.0, 0.3, 1.5, 31.3 pg/ml, respectively. Additionally, lung tissue homogenates were assessed for regulators of extracellular matrix deposition including matrix metalloproteinase (MMP)-3 and tissue inhibitor of metalloproteinase (TIMP)-1 (ELISA; R&D Systems; MDD of 0.125 and 0.031 ng/ml, respectively) as well as MMP-8 (ELISA; Abcam, Boston, MA; MDD of 0.053 ng/ml).
Lung cell staining and flow cytometry
Following removal of BALF and blood from pulmonary vasculature, harvested lungs (1/2 of right lungs) were subjected to an automated dissociation procedure using a gentleMACS Dissociator instrument (Miltenyi Biotech, Auburn, CA). Viability of total lung cells was assessed by trypan blue exclusion and a LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen, Carlsbad, CA). Cell viability was > 99% with no differences by treatment group(s) (data not shown). Lung cells were incubated with CD16/32 (Fc Block, BioLegend, San Diego, CA) to minimize non-specific antibody staining, then stained with monoclonal antibodies against rat anti-mouse; CD45 (clone: 30-F11; BD Biosciences, Franklin Lake, NJ), CD11b (clone: M1/70; BD Biosciences and BioLegend), Ly6G (clone: 1A8; BD Biosciences), CD11c (clone: N418; Invitrogen), CD4 (clone: RM4-5; BD Biosciences), CD8 (clone: 53–6.7; BD Biosciences), CD19 (clone: 1D3; Invitrogen), hamster anti-mouse CD3e (clone: 145-2C11; BD Biosciences and BioLegend), mouse anti-mouse NK1.1 (clone: PK136; BD Biosciences or BioLegend), Ly6C (clone: HK1.4; BioLegend), and F4/80 (clone: QA17A29; BioLegend or clone: T45-2342; BD Biosciences). Cells were acquired on a BD LSRII Yellow/Green cytometer configured with 355-nm, 405-nm, 488-nm, 561-nm, and 633-nm lasers. Post-acquisition, data were exported and stored using the flow cytometry standard (FCS) 3.1 format and analyzed using FlowJo software version 10.8 (FlowJo, RRID:SCR_008520, Ashland, OR).
The gating strategies for Ly6G + neutrophils, CD11c + CD11b lo alveolar (Alv) macrophages (Mɸ), CD11c + CD11b hi activated (Act) alveolar Mɸ, CD11c int CD11b hi recruited/transitioning monocytes-Mɸ, CD11c − CD11b hi monocytes, CD3 + CD4 + T cells, CD3 + CD8 + T cells, CD19 + B cells, and NK cells were performed as previously reported [ 12 , 17 , 18 ] with associated RFP + gating per cell population (Supplemental Fig. 1 and Fig. 1 ). The percentage of all respective cell populations was determined from live CD45 + lung leukocytes after excluding debris and doublets. This percentage was multiplied by the respective total lung cell numbers to determine specific cell population numbers for each animal.

Inhalation of organic dust extract (ODE), lipopolysaccharide (LPS), and peptidoglycan (PGN) induce lung CCR2 + monocyte-macrophages (Mɸ) and monocytes. CCR2 RFP/+ mice were exposed once to ODE (25%), LPS (10 μg), PGN (100 μg), or saline control and euthanized at 48 h. Scatter plots with bars depict mean with SD delineating cells as total (gray), CCR2 + (green), and CCR2 − (red). A Total lung cells enumerated. B Representative contour plot of the four monocyte (mono)-Mɸ subpopulations across groups based upon CD11c and CD11b expression after removal of neutrophils gated from live CD45 + cells after excluding debris and doublets. C RFP ± staining by exposure group and subpopulation. D CD11c + CD11b lo alveolar (Alv) Mɸ, CD11c + CD11b hi activated (Act) Mɸ, CD11c int CD11b hi mono-Mɸ, and CD11c − CD11b. hi monocytes determined by multiplying lung cell % population by total lung cells enumerated from lung sample. Statistical analyses were performed with Kruskal–Wallis with Dunn’s test for multiple comparisons (# p < 0.05, ## p < 0.01, ### p < 0.001, #### p < 0.0001) vs. respective saline. N = 19 (saline), 9 (ODE), 8 (LPS), 5 (PGN)
Lung histopathology and post-translational modifications
Following removal of BALF and blood from the pulmonary vasculature, left lungs were excised and inflated to 15 cm H 2 O pressure with 10% formalin (Fisher Scientific, Fair Lawn, NJ) for 24 h to preserve pulmonary architecture [ 18 ]. Fixed left lung lobes were then placed into cassettes, embedded in paraffin, cut (4–5 μm) at midpoint sections to include regions of both large and small airways as well as blood vessels, and stained with hematoxylin and eosin (H&E) or preserved for subsequent IHC. H&E-stained slides of entire lung sections from each animal were reviewed at all scanning magnifications and semi-quantitatively scored for the degree and distribution of lung inflammation. Scores were generated by an expert pathologist blinded to treatment conditions utilizing a previously published scoring system (scored 0 to 4) [ 18 , 33 ] that evaluates the spectrum of inflammatory changes for alveolar and bronchiolar compartments with higher scores indicating greater inflammation.
Lung sections were also stained with modified Masson’s Trichrome and scanned by Aperio scanner (Leica Biosystems, Deer Park, IL). The VENTANA image viewer (version 3.1.4; Roche Diagnostics, Indianapolis, IN) was utilized to capture 10 images per lung section at 20 × from scanned images. Collagen content in trichrome images was quantified as previously described [ 12 , 34 ] using Image J FIJI plugin (version: 2.9.0/1.53t U.S. National Institutes of Health, Bethesda, MD).
To quantify CCR2 + expression of inflammatory monocyte/macrophages, lung sections were stained with anti-CCR2 (1:100, NBP267700, Lot HM0537, Novus, Littleton, CO) and cross absorbed with donkey anti-rabbit (AlexaFluor488, A21206, Lot #2,156,521, Thermo Fisher, Waltham, MA) at 1:100 and processed as previously described [ 12 ]. Slides were mounted with VECTASHIELD® Antifade Mounting Medium with DAPI (4′6-diamindino-2-phenylindole; to identify nuclei)(Cat#H-1200, Lot#ZG1014, Curlingame, CA). Using a Zeiss fluorescent microscope (Zeiss Observer.Z1 Zeiss, White Plains, NY), photographs (10 lung images per mouse) of lung parenchyma were taken, and CCR2 + expression by integrated density was quantified by Image J FIJI plugin.
Citrullinated (CIT) and malondialdehyde acetaldehyde (MAA) modified proteins and vimentin were stained [ 17 ]. Increased in the context of inflammatory lung diseases, vimentin is an extracellular matrix protein that is also targeted by post-translational modifications generated during the process of inflammation and increased oxidative stress. Prior studies by our group have demonstrated robust co-localization of MAA and CIT with vimentin in lung tissues of mice and humans with inflammatory arthritis and lung disease [ 17 , 35 ]. Lung sections were stained with Cy5 rabbit anti-vimentin (Bioss, Woburn, MA, USA, 1:100), Zenon AF 594 label (Invitrogen, Carlsbad, CA, USA) and rabbit polyclonal IgG antibody to MAA [ 17 ], or a mouse monoclonal anti-peptidyl-citrulline antibody (clone F95 IgMκ, Millipore Sigma, Burlington, MA, USA). Detection of the F95 antibody was done using an AF 488 AffiniPure donkey anti-mouse IgM, µ chain specific antibody (Jackson Immunoresearch, West Grove, PA, USA). DAPI (4′,6-diamidino-2-phenylindole; to identify nuclei) was added and samples were sealed with Fluormount-G (Southern Biotech, Birmingham, AL, USA). Fluorochromes detected using a Revolve fluorescent microscope (ECHO, San Diego, CA, USA). Images were quantified using ImageJ, and colocalization was performed using the Image J (RRID:SCR_003070) FIJI plugin Coloc 2 [ 17 , 18 ].
Statistical analysis
Sample-size requirements were extrapolated from previous work assessing post-LPS lung recovery treatments in C57BL/6 [ 12 ]. The mean (± SD) of CD11c int CD11b hi transitioning/recruited monocyte-macrophages was 0.26 × 10 5 (0.09 × 10 5 ) with saline and 6.5 × 10 5 (2.2 × 10 5 ) with LPS treatment 48 h post-exposure; thus, a sample size of N = 2 in each group would achieve 80% power at the 0.05 level of significance to determine an influx of these cells following inflammatory agent exposure as compared to saline control. Experimental groups for the CCR2 trafficking studies include at least 2 mice for each group. The maximum sample sizes for the CCR2 RFP/+ trafficking studies are N = 19 (saline/Sal), N = 9 (ODE), N = 8 (LPS), and N = 5 (PGN). A sample size of n = 5 would achieve 80% power at the 0.05 level of significance to detect a 60% reduction in CD11c int CD11b hi transitioning/recruited monocyte-macrophages with depletion strategies (i.e., CCR2 KO and clodronate liposomes). For the CCR2 WT vs. KO studies, N = 5 (CCR2 WT-Sal), N = 5 (CCR2 KO-Sal), N = 9 (CCR2 WT-LPS) and N = 9 (CCR2 KO-LPS); and for the clodronate (Clod) versus vehicle (Veh) liposome targeted studies, N = 8 (Veh + Sal), 8 (Clod + Sal), 8 (Veh + LPS), and 9 (Clod + LPS). Experimental groups for those studies include at least 5 mice for each group. Numbers less than the maximum number reflect limitations in available sample quantity or quality.
Data are presented as the mean ± standard deviation (± SD) with scatter plots depicted for each data point. The Shapiro–Wilk test was utilized to test for normality among treatment groups. If the normality condition was satisfied, parametric statistical tests (one-way ANOVA with subsequent Tukey’s multiple comparison test), and if not satisfied, nonparametric statistical (Kruskal–Wallis with subsequent Dunn’s multiple comparison test) were used to assess differences between any two groups. All statistical analyses were performed using GraphPad Prism (version: 10.1.1) software and statistical significance accepted at a p value < 0.05.
Ethics statement
This study was conducted and reported in accordance with ARRIVE guidelines ( https://arriveguidelines.org ). All animal procedures were also approved by the University of Nebraska Medical Center (UNMC) Institutional Animal Care and Use Committee and were in accordance with the NIH guidelines for the use of rodents.
Inhalant exposures to organic dust extract (ODE), lipopolysaccharide (LPS), and peptidoglycan (PGN) induce lung infiltration of CCR2 + monocyte-macrophage (Mɸ) and monocyte subpopulations
In the first set of experiments, heterozygote CCR2 RFP/+ mice were treated once with ODE (25%), LPS (10 μg), PGN (100 μg), or sterile saline with lung tissue cell infiltrates analyzed at 48 h, as a previous study indicated that this was an optimal time point to detect recruited, infiltrating CD11c int CD11b hi transitioning monocytes-macrophages (Mɸ) following acute LPS treatment [ 12 ]. There were significant increases ( p < 0.05) in total cells, CCR2 + cells, and CCR2 − cells following ODE, LPS, and PGN as compared to saline control with no difference across the treatment groups (Fig. 1 A). The 4 monocyte-macrophage subpopulations including alveolar (Alv) Mɸ (CD11c + CD11b lo ), activated (Act) Mɸ (CD11c + CD11b hi ), transitioning monocyte-Mɸ (CD11c int CD11b hi ), and monocytes (CD11c − CD11b hi ) were delineated as previously described [ 12 ], with representative contour plots shown in Fig. 1 B. CCR2 RFP + and CCR2 RFP − expression in each of the four monocyte/Mɸ subpopulations by treatment condition are depicted in Fig. 1 C. CCR2 expression was absent on the Alv Mɸ and Act Mɸ subpopulations but was present on transitioning monocyte-Mɸ and monocyte subpopulations with numbers of these cell subpopulations enumerated in Fig. 1 D. The numbers of CCR2 + and CCR2 − transitioning monocyte-Mɸ were significantly increased with ODE, LPS, and PGN treatment as compared to saline control ( p < 0.05), but the magnitude of the increase was strikingly greater for the CCR2 + (vs. CCR2 − ) transitioning monocyte-Mɸ cells. There were also significant ( p < 0.05) increases in the numbers of CCR2 + and CCR2 − monocytes with ODE, LPS, and PGN treatment as compared to saline control with similar magnitude of increases for both CCR2 + and CCR2 − monocytes. CCR2 − Act Mɸ were increased with ODE, LPS, and PGN vs. saline, and correspondingly, CCR2 − Alv Mɸ were decreased with ODE, LPS, and PGN vs. saline (Fig. 1 D). Although differences vs. saline were demonstrated, there was no difference in the numbers of the monocytes/Mɸ among ODE, LPS, and PGN. Thus, all environmental exposures examined increased CCR2 + transitioning monocyte-Mɸ and monocyte subpopulations, but there were also increases in CCR2 − monocyte subpopulations and to a lesser degree CCR2 − transitioning monocyte-Mɸ.
Cell surface expression of Ly6C and F4/80 with monocyte/macrophage (Mɸ) subpopulations following inhalant exposures to ODE, LPS, and PGN
Cell surface expression of Ly6C, a predominant marker of monocytes and/or associated with pro-inflammatory and pro-fibrotic properties [ 36 ] by percent expression and mean fluorescence intensity (MFI) were also investigated and summarized (Fig. 2 ). Ly6C expression was low (< 5%) on Sal-Alv Mɸ and ODE-, LPS-, PGN-Act Mɸ (data not shown). In contrast, Ly6C percent and MFI expression were increased on both CCR2 + and CCR2 − ODE-, LPS-, PGN- induced CD11c int CD11b hi monocyte-Mɸ cells vs. saline control (except MFI expression was not increased for these CCR2 − cells following PGN exposure) (Fig. 2 A, B). Moreover, Ly6C MFI expression was significantly ( p < 0.05) increased on these CCR2 + monocyte-Mɸ cells associated with ODE, LPS, and PGN exposure as compared to the corresponding CCR2 − monocyte-Mɸ cells. Ly6C percent expression was high on all monocyte populations with a significant ( p < 0.05) increase with LPS-associated CCR2 + and CCR2 − monocytes vs. saline (Fig. 2 A, B). There was an increase in Ly6C MFI expression with ODE and LPS CCR2 − monocytes vs. saline with no difference in intensity of the MFI expression across CCR2 RFP + monocytes. As observed with transitioning monocyte-Mɸ cells, MFI expression was increased in all CCR2 + monocytes as compared to CCR2 − monocytes. These studies demonstrate that Ly6C expression was increased in the recruited CCR2 + cells as well as CCR2 − cells following exposure to environmental agents, and as such, Ly6C alone may not discriminate monocyte-macrophage subpopulations.

Ly6C expression of monocyte-macrophage (Mɸ) and monocyte subpopulations following organic dust extract (ODE), lipopolysaccharide (LPS), and peptidoglycan (PGN) exposure. C57BL/6 mice were exposed once to ODE (25%), LPS (10 μg), PGN (100 μg), or saline control and euthanized at 48 h. Scatter plots with bars depict mean with SD delineating cells as CCR2 + (green) and CCR2- (red). Expression of Ly6C by percent ( A ) and mean fluorescence intensity (MFI) ( B ) across CD11cintCD11bhi monocyte-Mɸ and CD11c-CD11b + monocyte subpopulations as determined by flow cytometry. Statistical analyses were performed with Kruskal–Wallis with Dunn’s test for multiple comparisons (# p < 0.05 vs. respective saline) and (* p < 0.05 denoted by line with brackets denoting difference between same inhalant exposure by CCR2 RFP positive vs. negative). N = 15 (saline), 9 (ODE), 4 (LPS), 5 (PGN)
The monocyte and macrophage marker F4/80 (ADGRE1) [ 37 ] was also investigated across cell subpopulations (Supplemental Fig. 2 ). The percent F4/80 expression was ubiquitous across Sal-Alv Mɸ and ODE-, LPS-, PGN-Act Mɸ subpopulations, but the expression intensity by MFI was increased in the ODE-, LPS-, PGN-Act Mɸ vs. Saline-Alv Mɸ (Supplemental Fig. 2 ). The percent F4/80 expression was high (~ 60–80%) in the transitioning CCR2 + and CCR2 − CD11c int CD11b hi monocyte-macrophage cells (~ 60–80%) and more variable with CD11c − CD11b hi monocytes.
Inhalant exposures to ODE and LPS induce lung infiltration of CCR2 +NK cells and T cells in addition to CCR2 − neutrophils, B cells, and T cells
The number of CCR2 + and CCR2 − neutrophils and lymphocytes was also investigated to capture any non-monocyte/macrophage cell-specific CCR2 expression events at 48 h post environmental agent exposure (Fig. 3 ). CCR2 is recognized to be expressed with NK cells [ 38 ] and activated T cells [ 39 ]. Indeed, there were significant ( p < 0.05) increases in CCR2 + NK cells, CD3 + CD4 + T cells, and CD3 + CD8 + T cells with ODE and LPS but not PGN treatment vs. saline. There were also significant ( p < 0.05) increases in CCR2 − CD3 + CD4 + T cells and CCR2 − CD3 + CD8 + T cells. ODE, LPS, and PGN treatment did not increase CCR2 + neutrophils or CD19 + B cells. ODE, LPS, and PGN treatment also increased CCR2 − neutrophils and CD4 + T cells, and ODE and LPS (but not PGN) increased CCR2 − CD8 + T cells.

Organic dust extract (ODE) and lipopolysaccharide (LPS) inhalation induce lung infiltration of CCR2 + NK and T cells. C57BL/6 mice were exposed once to ODE (25%), LPS (10 μg), PGN (100 μg), or saline control and euthanized at 48 h. Scatter plots with bars depict mean with SD delineating cells as total (gray), CCR2 + (green), and CCR2- (red). CD11c-Ly6G + neutrophils, CD19 + B cells, CD3-NK1.1 + NK cells, CD3 + CD4 + T cells, and CD3 + CD8 + T cell infiltrates determined by flow cytometry on live CD45 + cells after exclusion of debris and doublets with lung cell % populations multiplied by total lung cells enumerated from lung sample. Gating strategy depicted in Supplemental Fig. 1 . Statistical analyses were performed with Kruskal–Wallis with Dunn’s test for multiple comparisons (# p < 0.05, ## p < 0.01, ### p < 0.001, #### p < 0.0001) vs. respective saline. N = 19 (saline), 9 (ODE), 8 (LPS), 5 (PGN)
Inhalant LPS-induced lung inflammatory responses are not reduced in CCR2 knock-out (KO) mice
Because there were no major differences in ODE-, LPS-, and PGN-induced CCR2 + monocyte-macrophage lung cell infiltrates, LPS was utilized as the prototype environmental inflammatory agent for the remainder of the studies. It was hypothesized that CCR2 knock-out (KO) mice would be protected against LPS-induced lung inflammatory and pro-fibrotic responses due to reduction in the recruitment of transitioning CCR2 + monocyte-Mɸ infiltrates. Although there were significant ( p = 0.0005) reductions (54% reduction) in LPS-induced CD11c int CD11b hi monocyte-Mɸ cells, there were no significant reductions in LPS-induced pro-inflammatory and pro-fibrotic mediators in lung homogenates including TNF-α, IL-6, CXCL1, MMP-3, MMP-8, and TIMP-1 (Table 1 ) and lung histopathology (data not shown) in CCR2 KO mice vs. WT mice. There were also no differences between CCR2 KO and WT mice for LPS-induced total cells, neutrophils, lymphocytes, and macrophages in BALF as well as LPS-induced lung infiltrates including activated Mɸ, monocytes, T and B lymphocytes, and NK cells. In contrast, LPS-induced lung neutrophils were increased in CCR2 KO (vs. WT) mice, and moreover, there were corresponding, likely compensatory, increases in lung and serum CCL2 and CCL7, chemoattractants that predominately drive monocyte recruitment but also can affect lymphocytes and neutrophils, with the LPS treated CCR2 KO mice.
LPS-induced lung transitioning, infiltrating CD11c int CD11b hi are reduced with systemic delivery of clodronate liposomes
In an alternative approach to deplete recruited lung monocytes-macrophages induced by environmental exposures, intravenous clodronate liposomes (vs. vehicle control liposomes) were dosed one day prior to LPS (and saline) treatment to reduce circulating/systemic reservoir of available monocytes-macrophages with mice euthanized at 48 h following LPS exposure. In these studies, there was a reduction in total lung cells in tissue homogenates associated with a 59% reduction in LPS-induced lung CD11c int CD11b hi monocyte-Mɸ infiltrates in clodronate liposome pre-treated mice compared to control-treated with mice (Fig. 4 ). In contrast, there were no treatment differences in the number of LPS-induced Alv Mɸ, Act Mɸ or monocytes. There were also no treatment differences in the number of inflammatory cells in BALF and no treatment differences in the number of, CD19 + B cells, CD4 + and CD8 + T cells, and NK cells in tissue homogenates following LPS exposure (Supplemental Table 1 ).

LPS-induced transitioning CD11c int CD11b hi monocytes/macrophages are reduced with systemic delivery of clodronate liposomes. Mice were pre-treated with vehicle (Veh) or clodronate (Clod) liposomes 24 h prior to a one-time treatment with LPS (10 μg) or saline (Sal) control and euthanized at 48 h. Scatter plot with bars depicting mean with SD. A Total lung cells enumerated. B Representative contour plot of the four monocyte (mono)-Mɸ subpopulations across groups based upon CD11c and CD11b expression after removal of neutrophils gated from live CD45 + cells after excluding debris and doublets. C Number of CD11c + CD11b lo alveolar (Alv) Mɸ, CD11c + CD11b hi activated (Act) Mɸ, CD11c int CD11b hi mono-Mɸ, and CD11c − CD11b. hi monocytes determined by multiplying lung cell % population by total lung cells enumerated from lung sample. (# p < 0.05 vs. respective saline) and (* p < 0.05 denoted by line with brackets denoting difference between groups). N = 8 (Veh + Sal), 8 (Clod + Sal), 8 (Veh + LPS), 9 (Clod + LPS)
Effects of systemic delivery of clodronate liposomes with LPS-induced lung inflammation, collagen deposition, and infiltrating CCR2 + cells
Lung sections from these same mice pre-treated with vehicle and clodronate liposomes followed by saline and LPS challenge were evaluated for histopathological changes by H&E, collagen deposition by trichrome staining, and CCR2 + cell infiltrates (Fig. 5 A). Although semi-quantitative inflammatory scores following LPS exposure were reduced with clodronate liposome pre-treatment compared to vehicle control (Fig. 5 B), this difference did not reach statistical significance. LPS-induced collagen deposition was reduced by 23% ( p < 0.05) with clodronate liposome pretreatment (Fig. 5 C). Moreover, LPS-induced CCR2 + cell infiltrates were reduced by 60% ( p < 0.05) with clodronate liposome pretreatment (Fig. 5 D), consistent with reductions observed in CD11c int CD11b + monocyte- Mɸ demonstrated by flow cytometry.

Effects of systemic clodronate liposome delivery with LPS-induced lung inflammation, collagen, and infiltrating CCR2 + cells. Mice were pre-treated with vehicle (Veh) or clodronate (Clod) liposomes 24 h prior to a one-time treatment with LPS (10 μg) or saline (Sal) control and euthanized at 48 h. A Representative images from treatment groups stained by H&E, trichome, and CCR2 (red) with DAPI nuclei staining (blue) by confocal microscopy. Scatter plots with bars depict mean with SD of semi-quantitative lung inflammatory score ( B ) and integrated density of collagen ( C ), and CCR2 ( D ) quantified per each mouse. Statistical analyses were performed with Kruskal–Wallis with Dunn’s multiple comparison (inflammatory scores) and ANOVA with Tukey’s multiple comparison (collagen content and CCR2) (# p < 0.05 vs. respective saline) and (* p < 0.05 denoted by line with brackets denoting difference between groups). N = 8 (Veh + Sal), 8 (Clod + Sal), 8 (Veh + LPS), 9 (Clod + LPS). Line scale denotes 100 μm
LPS-induced lung pro-fibrotic and inflammatory mediators modulated following systemic delivery of clodronate liposomes
Pre-treatment with intravenous clodronate liposomes (vs. vehicle) also resulted in significant reductions in LPS-induced levels of pro-fibrotic mediators in lung homogenates including MMP-3 (33% reduction), MMP-8 (50% reduction), TIMP-1 (64% reduction), and TGF-β (38% reduction) (Fig. 6 ). Moreover, there were also significant ( p < 0.05) reductions in LPS-induced pro-inflammatory mediators including IL-6 (72% reduction) and neutrophil chemoattractant CXCL1 (57% reduction) with clodronate (vs. vehicle) liposome pre-treatment (Fig. 6 ). Lung levels of TNF-α induced by LPS exposure were not reduced with clodronate liposome pre-treatment, and there were also no differences in LPS-induced lung CCL2 and CCL7 levels between clodronate and vehicle liposome pre-treatment (Supplemental Table 1 ).

LPS-induced lung pro-fibrotic and inflammatory mediators modulated following systemic delivery of clodronate liposomes. Mice were pre-treated with vehicle (Veh) or clodronate (Clod) liposomes 24 h prior to a one-time treatment with LPS (10 μg) or saline (Sal) control and euthanized at 48 h. Scatter plots with bars depict mean with SD of protein levels of matrix metalloproteinase (MMP)-3, MMP-8, metalloproteinase inhibitor (TIMP-1), transforming growth factor (TGF)-β, IL-6, and neutrophil chemokine CXCL1 of lung homogenate. Statistical analyses were performed with ANOVA and Tukey’s multiple comparison test with significance (# p < 0.05 vs. respective saline) and (* p < 0.05 denoted by line with brackets denoting difference between groups) with % reduction noted. N = 8 (Veh + Sal), 8 (Clod + Sal), 8 (Veh + LPS), 9 (Clod + LPS)
LPS-induced lung CIT and MAA autoantigens and vimentin expression were reduced with systemic delivery of clodronate liposomes
Based upon findings of decreased pro-fibrotic mediators and prior findings demonstrating that repetitive inhalant environmental exposures induce post-translational changes implicated in inflammatory and fibrotic lung disease [ 17 , 18 ], lung tissues were stained for CIT- and MAA-modified antigens as well as vimentin. CIT- and MAA-modified proteins and vimentin were significantly increased following a one-time LPS exposure vs. saline control at 48 h post-LPS exposure (Fig. 7 A, B). Moreover, there were significant ( p < 0.05) reductions in LPS-induced lung CIT-modified protein expression (39% reduction), MAA-modified protein expression (48% reduction), and vimentin (40% reduction) with clodronate (vs. vehicle) liposome administration.

LPS-induced lung CIT and MAA autoantigens and vimentin expression decrease with systemic clodronate liposome delivery. Mice were pre-treated with vehicle (Veh) or clodronate (Clod) liposomes 24 h prior to a one-time treatment with LPS (10 μg) or saline (Sal) control and euthanized at 48 h. A Representative confocal microscopy images of lung tissue from treatment groups stained for citrulline (CIT, green) and malondialdehyde-acetaldehyde (MAA, red) modified proteins and vimentin (teal). Line scale denotes 70 μm. B Scatter plots with bars depict mean with SD of integrated density of CIT- and MAA-modified proteins and vimentin quantified per each mouse. Statistical analyses were performed with ANOVA with Tukey’s for multiple comparisons (# p < 0.05 vs. respective saline) and (* p < 0.05 denoted by line with brackets denoting difference between groups). N = 5 (Veh + Sal), 7 (Clod + Sal), 8 (Veh + LPS), 9 (Clod + LPS)
Lung disease represents a major cause of occupation-related illness for which therapeutic approaches to alleviate disease burden are lacking. Recruited, infiltrating, and transitioning monocytes-macrophages have been implicated as critical cells in the immunopathogenesis of chronic lung disease. Here, our preclinical animal studies first defined and compared the trafficking of the inflammatory CCR2 + monocytes/macrophages to the lung following organic dust, endotoxin, and peptidoglycan exposure. These studies demonstrated striking increases in CCR2 + recruited/transitioning CD11c int CD11b hi monocyte-Mɸ and CD11c − CD11b hi monocyte subpopulations as well as striking increases in CCR2 − monocytes with these environmental exposures. However, CCR2 KO mice were not protected against inflammatory responses induced following endotoxin exposure despite a reduction (54%) in exposure induced CCR2 + recruited/transitioning CD11c int CD11b hi cells. Instead, systemic depletion of monocytes by intravenous clodronate liposome administration was associated with not only a reduction (59% reduction) in these transitioning monocyte-macrophages, but also a corresponding reduction in endotoxin-induced collagen deposition, extracellular matrix release, vimentin, and lung autoantigen expression. Thus, preventing the influx of circulating/recruited monocytes to the lung, as opposed to specifically targeting CCR2, following environmental exposures may represent a strategic area to further develop to reduce disease burden in occupationally exposed at-risk persons.
Occupational and environmental exposures are inherently complex, with highly concentrated exposures commonplace in a variety of settings, which can initiate and perpetuate the development of lung disease. In our studies aimed at defining the trafficking of monocytes-macrophages to the lung, we observed similar effects following separate exposures to high concentrations of complex organic dust extract (ODE), LPS, and PGN, all of which resulted in a robust increase in monocyte-macrophage recruitment. This supports the current focus of therapeutic approaches for environmental and occupational-associated diseases that have centered on specific agent identification and risk reduction measures and mitigation [ 40 ]. Supporting the relevance of experiments focused on high exposure doses, endotoxin levels encountered in real-world settings are highly variable and often exceed occupational exposure limits in agriculture settings by several orders of magnitude [ 41 , 42 ].
Lung monocytes-macrophages are important in mediating the response to inflammatory bioaerosol exposure, and recruited/transitional monocytes-macrophages specifically are implicated in the transition of acute inflammation to lung fibrosis in animal models and clinical investigations [ 43 ]. CCR2-expressing leukocytes are required for the progression of bleomycin-induced fibrosis, as CCR2 KO mice were protected against collagen deposition, macrophage infiltration, and MMP deposition in this model [ 44 , 45 ]. Likewise, pre-treatment with a CCR2 antagonist reduced lung fibrosis in a mouse model of scleroderma [ 46 ]. Although lung injury promotes the release of CCL2 by several different lung cell types, including airway epithelial cells, and serves as a chemoattractant for CCR2 pro-fibrotic macrophages [ 47 , 48 ], CCL2 neutralizing antibodies did not alter disease progression or mitigate lung function decline in patients with idiopathic pulmonary fibrosis, but instead was associated with an increase in endogenous CCL2 expression and other adverse compensatory changes [ 49 ]. We also found that CCL2 levels were increased in LPS-exposed CCR2 KO mice. The lack of efficacy observed with CCL2 targeting is consistent with our results in which CCL2 KO mice did not appear to be meaningfully protected from LPS-induced lung injury.
In endotoxin exposure models of lung injury, several studies have also demonstrated that there is a decrease in the endotoxin-induced recruitment of peripheral blood monocytes, exudative macrophages, F4/80 + lung cells, lung neutrophils, and lung cell infiltrates in CCR2 KO mice as well as mice depleted of systemic monocytes by clodronate liposomes [ 25 , 50 , 51 , 52 , 53 , 54 ]. Our studies confirm the reduction in infiltrating monocytes/macrophages with both the CCR2 KO and the administration clodronate liposomes, but also simultaneously highlight striking differences in these modeling approaches to reduce profibrotic and inflammatory responses following LPS exposure. In addition to finding no meaningful benefit for globally depleting CCR2-expressing cells in endotoxin-induced lung disease, we demonstrated that CCR2 KO mice had increased neutrophil accumulation with compensatory increases in CCL2 and CCL7 following LPS exposure, suggesting this strategy may actually yield detrimental effects.
Our observations are consistent with a study by Gurczynski and colleagues demonstrating no protection against ɣ-herpesvirus-induced pneumonitis and fibrosis with CCR2 KO mice and that CCR2 + cells played a suppressive role by limiting collagen and IL-17 production [ 55 ]. Another potential compensatory mechanism supported by this study is that the environmental exposures induced the expression of CCR2 + on the CD4 + and CD8 + T-cell infiltrates. Others have demonstrated suppressive roles for CCR2-expressing T cells in lung infections and inflammatory responses [ 56 ]. Moreover, CCR2 is highly expressed on NK cells, and both ODE and LPS induced the recruitment of NK cells. Thus, depletion of CCR2 function with lymphocytes may have negated any potential benefit of reducing CCR2 + monocytes-macrophages. Based on these findings, further investigations are warranted to understand the functional role of CCR2 on infiltrating lymphocytes in the setting of environmental exposure-induced lung disease.
In contrast to CCR2 targeting, the strategy of inhibiting the recruitment of peripheral blood monocytes to the lung through the systemic administration of clodronate liposomes demonstrated benefit with most, but not all, endpoints examined. Beneficial responses are best characterized by a reduction in the pro-fibrotic properties of lung monocytes-macrophages. Namely, endotoxin-induced collagen deposition, MMP3, MMP8, TIMP-1, TGF-β, IL-6, and CXCL1 (but not TNF-⍺) were reduced by depleting peripheral blood monocytes. This corresponded with reductions observed in CIT- and MAA-modified protein generation following endotoxin exposure. This is relevant, as proteins co-modified with CIT and MAA stimulate undifferentiated macrophages towards a mixed M1/M2 phenotype [ 57 ] and secrete soluble factors that drive an aggressive fibroblast phenotype [ 20 ]. Whereas there was no compensatory increase in CCL2 or CCL7 release following the administration of clodronate liposomes as observed with CCR2 KO mice, neither strategy appeared to mitigate the histopathologic changes initiated by acute LPS exposure.
These collective findings underscore the importance of the recruited peripheral blood monocyte transitioning to a lung monocyte/macrophage population in mediating pro-fibrotic processes in the lung as well as generation of post-translationally modified proteins following endotoxin exposure. Therefore, depletion and/or inhibition of “recruitable” peripheral blood monocytes may represent a novel strategy to reduce the burden of lung diseases resulting from select environmental exposures. Whereas CCR2 abolition did not exhibit protection against LPS-induced inflammatory or pro-fibrotic responses, antagonism of other monocyte trafficking receptors (i.e., CCR1, 5, 6, 7), blockade of adhesion molecules (i.e., selectins, integrins, ICAM-1/VCAM-1), or inhibition of chemokines (i.e., CCL2, CCL5, CCL7) may demonstrate therapeutic benefit given depletion of the recruitable monocyte reservoir demonstrated protective effects in our model of acute exposure-induced lung inflammation [ 58 , 59 , 60 ]. Moreover, understanding the mechanisms governing the crosstalk between lung monocytes and airway structural cells including fibroblasts/myofibroblasts is warranted.
In conclusion, high concentration exposure to environmental and occupational exposures including agricultural organic dust extracts, endotoxin, and peptidoglycan induce the recruitment of CCR2 + and CCR2 − peripheral blood monocytes transitioning to lung resident monocytes/macrophages. Depleting peripheral blood monocytes by systemic administration of clodronate liposomes, but not through CCR2 KO animal strategies, resulted in the reduction of endotoxin-induced pro-inflammatory and pro-fibrotic mediators. Developing translational strategies to reduce the recruitment of these cells following exposures may be warranted to reduce risk of lung disease.
Availability of data and materials
Data that support the findings of this study have been deposited in Zenodo. The data are embargoed until manuscript acceptance for publication. The link to the data is below. https://zenodo.org/records/10641513?token=eyJhbGciOiJIUzUxMiJ9.eyJpZCI6IjNjYmNhNDA0LThmMDYtNDBmZS1iMTc2LTZkYTQ1NDZjZjExOCIsImRhdGEiOnt9LCJyYW5kb20iOiIzODkyNWQ0MmUxYzdlNzE4Zjg5YWRkZDVhMzllMTYzNiJ9.KUYVheTKH7IITATY9EiCchsVBH4Db6OoaIi-c2fAakjMpTypBhSTuAHeXMnz-9qoeyVrUzXboA0T7EpsPGMwVw .
Seaman DM, Meyer CA, Kanne JP. Occupational and environmental lung disease. Clin Chest Med. 2015;36(2):249–68 viii-ix.
Article PubMed Google Scholar
Després V, Huffman JA, Burrows SM, Hoose C, Safatov A, Buryak G, et al. Primary biological aerosol particles in the atmosphere: a review. Tellus B Chem Phys Meteorol. 2012;64(1):15598.
Article Google Scholar
Park Y, Ahn C, Kim TH. Occupational and environmental risk factors of idiopathic pulmonary fibrosis: a systematic review and meta-analyses. Sci Rep. 2021;11(1):4318.
Article CAS PubMed PubMed Central Google Scholar
Schwab AD, Poole JA. Mechanistic and therapeutic approaches to occupational exposure-associated allergic and non-allergic asthmatic disease. Curr Allergy Asthma Rep. 2023;23(6):313–24.
Lee CT, Feary J, Johannson KA. Environmental and occupational exposures in interstitial lung disease. Curr Opin Pulm Med. 2022;28(5):414–20.
Article CAS PubMed Google Scholar
Blanc PD, Annesi-Maesano I, Balmes JR, Cummings KJ, Fishwick D, Miedinger D, et al. The occupational burden of nonmalignant respiratory diseases. An official American thoracic society and european respiratory society statement. Am J Respir Crit Care Med. 2019;199(11):1312–34.
Article PubMed PubMed Central Google Scholar
Perlman DM, Maier LA. Occupational lung disease. Med Clin North Am. 2019;103(3):535–48.
Collaborators GOCRRF. Global and regional burden of chronic respiratory disease in 2016 arising from non-infectious airborne occupational exposures: a systematic analysis for the Global Burden of Disease Study 2016. Occup Environ Med. 2020;77(3):142–50.
Collaborators GOCRRF. Global burden of chronic respiratory diseases and risk factors, 1990–2019: an update from the global burden of disease study 2019. EClinicalMedicine. 2023;59:101936.
Basinas I, Sigsgaard T, Kromhout H, Heederik D, Wouters IM, Schlunssen V. A comprehensive review of levels and determinants of personal exposure to dust and endotoxin in livestock farming. J Expo Sci Environ Epidemiol. 2015;25(2):123–37.
Moller W, Heimbeck I, Hofer TP, Khadem Saba G, Neiswirth M, Frankenberger M, et al. Differential inflammatory response to inhaled lipopolysaccharide targeted either to the airways or the alveoli in man. PLoS ONE. 2012;7(4):e33505.
Poole JA, Gaurav R, Schwab A, Nelson AJ, Gleason A, Romberger DJ, et al. Post-endotoxin exposure-induced lung inflammation and resolution consequences beneficially impacted by lung-delivered IL-10 therapy. Sci Rep. 2022;12(1):17338.
Rolph CA, Gwyther CL, Tyrrel SF, Nasir ZA, Drew GH, Jackson SK, et al. Sources of airborne endotoxins in ambient air and exposure of nearby communities—a review. Atmosphere. 2018;9(10):375.
Article CAS Google Scholar
Tager IB, Lurmann FW, Haight T, Alcorn S, Penfold B, Hammond SK. Temporal and spatial patterns of ambient endotoxin concentrations in Fresno, California. Environ Health Perspect. 2010;118(10):1490–6.
Park S, Allen RJ, Lim CH. A likely increase in fine particulate matter and premature mortality under future climate change. Air Qual Atmos Health. 2020;13(2):143–51.
Joshi M, Goraya H, Joshi A, Bartter T. Climate change and respiratory diseases: a 2020 perspective. Curr Opin Pulm Med. 2020;26(2):119–27.
Poole JA, Mikuls TR, Thiele GM, Gaurav R, Nelson AJ, Duryee MJ, et al. Increased susceptibility to organic dust exposure-induced inflammatory lung disease with enhanced rheumatoid arthritis-associated autoantigen expression in HLA-DR4 transgenic mice. Respir Res. 2022;23(1):160.
Mikuls TR, Gaurav R, Thiele GM, England BR, Wolfe MG, Shaw BP, et al. The impact of airborne endotoxin exposure on rheumatoid arthritis-related joint damage, autoantigen expression, autoimmunity, and lung disease. Int Immunopharmacol. 2021;100:108069.
Poole JA, Thiele GM, Ramler E, Nelson AJ, Duryee MJ, Schwab AD, et al. Combined repetitive inhalant endotoxin and collagen-induced arthritis drives inflammatory lung disease and arthritis severity in a testosterone-dependent manner. Am J Physiol Lung Cell Mol Physiol. 2024;326:L239–L251.
Aripova N, Duryee MJ, England BR, Hunter CD, Mordeson JE, Ryan EM, et al. Citrullinated and malondialdehyde-acetaldehyde modified fibrinogen activates macrophages and promotes an aggressive synovial fibroblast phenotype in patients with rheumatoid arthritis. Front Immunol. 2023;14:1203548.
Warren KJ, Wyatt TA, Romberger DJ, Ailts I, West WW, Nelson AJ, et al. Post-injury and resolution response to repetitive inhalation exposure to agricultural organic dust in mice. Safety (Basel). 2017;3(1):10.
Laskin DL, Malaviya R, Laskin JD. Role of macrophages in acute lung injury and chronic fibrosis induced by pulmonary toxicants. Toxicol Sci. 2019;168(2):287–301.
Okabe Y, Medzhitov R. Tissue biology perspective on macrophages. Nat Immunol. 2016;17(1):9–17.
David BA, Kubes P. Exploring the complex role of chemokines and chemoattractants in vivo on leukocyte dynamics. Immunol Rev. 2019;289(1):9–30.
Oliveira VLS, Pollenus E, Berghmans N, Queiroz-Junior CM, Blanter M, Mattos MS, et al. Absence of CCR2 promotes proliferation of alveolar macrophages that control lung inflammation in acute respiratory distress syndrome in mice. Int J Mol Sci. 2022;23(21):12920.
Poole JA, Dooley GP, Saito R, Burrell AM, Bailey KL, Romberger DJ, et al. Muramic acid, endotoxin, 3-hydroxy fatty acids, and ergosterol content explain monocyte and epithelial cell inflammatory responses to agricultural dusts. J Toxicol Environ Health Part A. 2010;73(10):684–700.
Nelson AJ, Roy SK, Warren K, Janike K, Thiele GM, Mikuls TR, et al. Sex differences impact the lung-bone inflammatory response to repetitive inhalant lipopolysaccharide exposures in mice. J Immunotoxicol. 2018;15(1):73–81.
Poole JA, Gleason AM, Bauer C, West WW, Alexis N, Reynolds SJ, et al. alphabeta T cells and a mixed Th1/Th17 response are important in organic dust-induced airway disease. Ann Allergy Asthma Immunol. 2012;109(4):266–73.e2.
Tatham KC, O’Dea KP, Romano R, Donaldson HE, Wakabayashi K, Patel BV, et al. Intravascular donor monocytes play a central role in lung transplant ischaemia-reperfusion injury. Thorax. 2018;73(4):350–60.
Zhang L, Xie W, Zhang J, Shanahan H, Tonello R, Lee SH, et al. Key role of CCR2-expressing macrophages in a mouse model of low back pain and radiculopathy. Brain Behav Immun. 2021;91:556–67.
Zito MA, Koennecke LA, McAuliffe MJ, McNally B, van Rooijen N, Heyes MP. Depletion of systemic macrophages by liposome-encapsulated clodronate attenuates striatal macrophage invasion and neurodegeneration following local endotoxin infusion in gerbils. Brain Res. 2001;892(1):13–26.
Ribeiro-Machado C, Santos SG, Amaral IA, Caldeira J, Pereira P, Barbosa MA, et al. Macrophage-based therapy for intervertebral disc herniation: preclinical proof-of-concept. NPJ Regen Med. 2023;8(1):34.
Tantawy MA, Hatesuer B, Wilk E, Dengler L, Kasnitz N, Weiß S, et al. The interferon-induced gene Ifi27l2a is active in lung macrophages and lymphocytes after influenza A infection but deletion of Ifi27l2a in mice does not increase susceptibility to infection. PLoS ONE. 2014;9(9):e106392.
Chen Y, Yu Q, Xu C-B. A convenient method for quantifying collagen fibers in atherosclerotic lesions by ImageJ software. Int J Clin Exp Med. 2017;10(10):14904–10.
Google Scholar
England BR, Duryee MJ, Roul P, Mahajan TD, Singh N, Poole JA, et al. Malondialdehyde-acetaldehyde adducts and antibody responses in rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. 2019;71(9):1483–93.
Gibbons MA, Mackinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated pro-fibrotic macrophage regulation of lung fibrosis. Am J Respir Crit Care Med. 2011;184:569–81.
Waddell LA, Lefevre L, Bush SJ, Raper A, Young R, Lisowski ZM, et al. ADGRE1 (EMR1, F4/80) is a rapidly-evolving gene expressed in mammalian monocyte-macrophages. Front Immunol. 2018;9:2246.
Fujimura N, Xu B, Dalman J, Deng H, Aoyama K, Dalman RL. CCR2 inhibition sequesters multiple subsets of leukocytes in the bone marrow. Sci Rep. 2015;5:11664.
Bakos E, Thaiss CA, Kramer MP, Cohen S, Radomir L, Orr I, et al. CCR2 Regulates the immune response by modulating the interconversion and function of effector and regulatory T cells. J Immunol. 2017;198(12):4659–71.
Vlahovich KP, Sood A. A 2019 update on occupational lung diseases: a narrative review. Pulm Ther. 2021;7(1):75–87.
Davidson ME, Schaeffer J, Clark ML, Magzamen S, Brooks EJ, Keefe TJ, et al. Personal exposure of dairy workers to dust, endotoxin, muramic acid, ergosterol, and ammonia on large-scale dairies in the high plains Western United States. J Occup Environ Hyg. 2018;15(3):182–93.
Roque K, Shin KM, Jo JH, Lim GD, Song ES, Shin SJ, et al. Association between endotoxin levels in dust from indoor swine housing environments and the immune responses of pigs. J Vet Sci. 2018;19(3):331–8.
Ogawa T, Shichino S, Ueha S, Matsushima K. Macrophages in lung fibrosis. Int Immunol. 2021;33(12):665–71.
Baran CP, Opalek JM, McMaken S, Newland CA, O’Brien JM Jr, Hunter MG, et al. Important roles for macrophage colony-stimulating factor, CC chemokine ligand 2, and mononuclear phagocytes in the pathogenesis of pulmonary fibrosis. Am J Respir Crit Care Med. 2007;176(1):78–89.
Okuma T, Terasaki Y, Kaikita K, Kobayashi H, Kuziel WA, Kawasuji M, et al. C-C chemokine receptor 2 (CCR2) deficiency improves bleomycin-induced pulmonary fibrosis by attenuation of both macrophage infiltration and production of macrophage-derived matrix metalloproteinases. J Pathol. 2004;204(5):594–604.
Ishikawa M, Yamamoto T. Antifibrogenic effects of C-C chemokine receptor type 2 antagonist in a bleomycin-induced scleroderma model. Exp Dermatol. 2021;30(1):179–84.
Osterholzer JJ, Olszewski MA, Murdock BJ, Chen GH, Erb-Downward JR, Subbotina N, et al. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J Immunol. 2013;190(7):3447–57.
Yang J, Velikoff M, Canalis E, Horowitz JC, Kim KK. Activated alveolar epithelial cells initiate fibrosis through autocrine and paracrine secretion of connective tissue growth factor. Am J Physiol Lung Cell Mol Physiol. 2014;306(8):L786–96.
Raghu G, Martinez FJ, Brown KK, Costabel U, Cottin V, Wells AU, et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur Respir J. 2015;46(6):1740–50.
Dhaliwal K, Scholefield E, Ferenbach D, Gibbons M, Duffin R, Dorward DA, et al. Monocytes control second-phase neutrophil emigration in established lipopolysaccharide-induced murine lung injury. Am J Respir Crit Care Med. 2012;186(6):514–24.
Maus UA, Waelsch K, Kuziel WA, Delbeck T, Mack M, Blackwell TS, et al. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol. 2003;170(6):3273–8.
Cui TX, Brady AE, Fulton CT, Zhang YJ, Rosenbloom LM, Goldsmith AM, et al. CCR2 mediates chronic LPS-induced pulmonary inflammation and hypoalveolarization in a murine model of bronchopulmonary dysplasia. Front Immunol. 2020;11:579628.
Nakano H, Lyons-Cohen MR, Whitehead GS, Nakano K, Cook DN. Distinct functions of CXCR4, CCR2, and CX3CR1 direct dendritic cell precursors from the bone marrow to the lung. J Leukoc Biol. 2017;101(5):1143–53.
Rosseau S, Hammerl P, Maus U, Walmrath HD, Schutte H, Grimminger F, et al. Phenotypic characterization of alveolar monocyte recruitment in acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol. 2000;279(1):L25–35.
Gurczynski SJ, Procario MC, O’Dwyer DN, Wilke CA, Moore BB. Loss of CCR2 signaling alters leukocyte recruitment and exacerbates gamma-herpesvirus-induced pneumonitis and fibrosis following bone marrow transplantation. Am J Physiol Lung Cell Mol Physiol. 2016;311(3):L611–27.
Milger K, Yu Y, Brudy E, Irmler M, Skapenko A, Mayinger M, et al. Pulmonary CCR2(+)CD4(+) T cells are immune regulatory and attenuate lung fibrosis development. Thorax. 2017;72(11):1007–20.
Aripova N, Duryee MJ, Hunter CD, Ryan EM, Daubach EC, Jones SQ, et al. Peptidyl arginine deiminase expression and macrophage polarization following stimulation with citrullinated and malondialdehyde-acetaldehyde modified fibrinogen. Int Immunopharmacol. 2022;110:109010.
Belchamber KBR, Hughes MJ, Spittle DA, Walker EM, Sapey E. New Pharmacological Tools to Target Leukocyte Trafficking in Lung Disease. Front Immunol. 2021;12:704173.
Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. 2015;107(3):321–30.
Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11(11):762–74.
Download references
Acknowledgements
The authors acknowledge Craig Semerad, Victoria B. Smith, and Holly Britton in the Flow Cytometry Research Core Facility at the University of Nebraska Medical Center for aiding with flow cytometry studies. This core facility is administrated through the Office of the Vice Chancellor for Research and supported by state funds from the Nebraska Research Initiative (NRI) and The Fred and Pamela Buffett Cancer Center's National Cancer Institute Cancer Support Grant. Major instrumentation has been provided by the Office of the Vice Chancellor for Research, The University of Nebraska Foundation, the Nebraska Banker's Fund, and by the NIH-NCRR Shared Instrument Program. The authors would also like to thank Marie Nguyen for assistance in manuscript preparation and submission.
Disclosures
JAP has received research reagent from AstraZeneca (no monies) and has been a site investigator for allergy and asthma clinical studies for Takeda, GlaxoSmithKline, Regeneron, Areteia, and AstraZeneca (no monies). TRM received research support from Horizon Therapeutics and has been a consultant for Horizon, Pfizer, UCB, and Sanofi.
National Institute for Occupational Safety and Health grant U54OH010162 (ADS, TAW) and R01OH012045 (JAP), Department of Defense #PR200793 (JAP and TRM). ADS, TAW, and JAP received support from Central States Center of Agricultural Safety and Health (CS-CASH). TAW is supported by grants from the VA (I01 BX005886) and National Institutes of Health (P50 AA030407). TAW is the recipient of a Research Career Scientist Award (IK6 BX005962) from the Department of Veterans Affairs. TRM is also funded by VA Merit I01 BX004660.
Author information
Authors and affiliations.
Division of Allergy & Immunology, University of Nebraska Medical Center, Omaha, NE, USA
Aaron D. Schwab, Grace Moravec, Amy J. Nelson, Angela Gleason, Oliver Schanze & Jill A. Poole
Veterans Affairs Nebraska-Western Iowa Health Care System, Research Service, Omaha, NE, USA
Todd A. Wyatt, Geoffrey M. Thiele, Michael J. Duryee, Debra J. Romberger & Ted R. Mikuls
Division of Pulmonary, Critical Care & Sleep, University of Nebraska Medical Center, Omaha, NE, USA
Todd A. Wyatt & Debra J. Romberger
Department of Environmental, Agricultural and Occupational Health, College of Public Health, University of Nebraska Medical Center, Omaha, NE, USA
Todd A. Wyatt
Division of Rheumatology and Immunology, Department of Internal Medicine, College of Medicine, University of Nebraska Medical Center, Omaha, NE, USA
Geoffrey M. Thiele, Michael J. Duryee & Ted R. Mikuls
You can also search for this author in PubMed Google Scholar
Contributions
Conceived and designed research: JAP, TRM, GMT; performed experiments: JAP, TRM, GMT, AJN, MJD, ADS, AG, OS, GM, TAW; analyzed data: JAP, TRM, GMT, AJN, MJD, ADS, AG, OS, GM, TAW; interpreted results of experiments: JAP, TRM, GMT, AJN, MJD, ADS, AG, OS, GM, TAW; prepared figures: JAP, TRM, GMT, AJN, MJD, ADS; drafted manuscript: JAP, ADS; edited and revised manuscript: JAP, TRM, GMT, AJN, MJD, ADS, AG, OS, GM, TAW, DJM; approved final version of manuscript: JAP, TRM, GMT, AJN, MJD, ADS, AG, OS, GM, TAW, DJM.
Corresponding author
Correspondence to Jill A. Poole .
Ethics declarations
Ethics approval and consent to participate.
Neither human participants, data, nor tissues were used in these studies. The study was conducted and reported in accordance with ARRIVE guidelines ( https://arriveguidelines.org ). All animal procedures were approved by the University of Nebraska Medical Center (UNMC) Institutional Animal Care and Use Committee (IACUC) and were in accordance with NIH guidelines for the use of rodents.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: supplemental fig. 1..
Gating strategy for identification of non-debris, singlets, live CD45 + myeloid and lymphoid cells. For flow analysis, all panels were first gated as forward scatter-area (FSC-A) x side scatter-area (SSC-A) to omit debris, dead, or apoptotic cells. This was followed by two single cell gates to omit doublets (FSC-A x FSC-heigh (H) and SSC-A x SSC-H), followed by live/dead gate and then CD45 gate to assure removal of any additional dead or apoptotic cells and non-leukocytes. The CD45 + cells were placed on a CD11c x Ly6G gate to select Ly6G + neutrophils. Non-neutrophils were gated for CD19 + B cells (CD19 x SSC gate). This was followed by non-B cells gated on CD11c x CD11b gate to select CD11c + CD11b lo alveolar (Alv) macrophages (Mɸ), CD11c + CD11b hi activated (act) Mɸ, CD11c int CD11b hi transitioning monocytes (Mono)—Mɸ, and CD11c − CD11b hi monocytes (Mono). The negative or non-monocyte/macrophage populations were placed on CD3 x NK1.1 to select CD3 + T cells and CD3 − NK1.1 + NK cells, and then a CD4 x CD8 gate to select CD3 + CD4 + and CD3 + CD8 + T cells. A CCR2 RFP x SSC gate is shown for neutrophils and lymphocytes to demonstrate CCR2 + staining on specific lung cell subpopulations. Lung sample shown is from an LPS-exposed mouse.
Additional file 2: Supplemental Fig. 2.
F4/80 (ADGRE1) expression on monocyte-macrophage (Mɸ) subpopulations following organic dust extract (ODE), lipopolysaccharide (LPS), and peptidoglycan (PGN) inhalation exposure. C57BL/6 mice were exposed once to ODE (25%), LPS (10 μg), PGN (100 μg), or saline control and euthanized at 48 h. Scatter plots with bars depict mean with SD delineating cells as CCR2 + (green) and CCR2 − (red). Expression of F4/80 by percent (A) and mean fluorescence intensity (MFI) (B) across alveolar (Alv) Mɸ, activated (Act) Mɸ, monocyte-Mɸ, and monocyte subpopulations as determined by flow cytometry. Statistical analyses were performed with Kruskal–Wallis with Dunn’s test for multiple comparisons (# p < 0.05 vs. respective saline) and (* p < 0.05 denoted by line with brackets denoting difference between same inhalant exposure by CCR2 RFP positive vs. negative). N = 15 (saline), 9 (ODE), 4 (LPS), 5 (PGN).
Additional file 3: Supplemental Table 1.
LPS-induced airway inflammatory indices not affected with systemic delivery of clodronate liposomes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Schwab, A.D., Wyatt, T.A., Moravec, G. et al. Targeting transitioning lung monocytes/macrophages as treatment strategies in lung disease related to environmental exposures. Respir Res 25 , 157 (2024). https://doi.org/10.1186/s12931-024-02804-3
Download citation
Received : 09 February 2024
Accepted : 03 April 2024
Published : 09 April 2024
DOI : https://doi.org/10.1186/s12931-024-02804-3
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Organic dust
- Inflammation
- Lung disease
Respiratory Research
ISSN: 1465-993X
- General enquiries: [email protected]
IMAGES
VIDEO
COMMENTS
1. Introduction. Liposomes are self-assembled (phospho)lipid-based drug vesicles that form a bilayer (uni-lamellar) and/or a concentric series of multiple bilayers (multilamellar) enclosing a central aqueous compartment [].The size of liposomes ranges from 30 nm to the micrometer scale, with the phospholipidbilayer being 4-5 nm thick [].The field of liposomology was launched by the British ...
Many liposomal-based drug delivery systems are currently clinically approved to treat several diseases, such as cancer, fungal and viral infections; more liposomes have reached advanced phases in clinical trials. This review describes liposomes structure, composition, preparation methods, and clinical applications. 1.
To date, liposomes have been investigated in several pharmaceutical research as drug delivery systems and continue to constitute an intense field of research (Bozzuto and Molinari, 2015).Liposomes are considered a powerful drug delivery systems due to their structural versatility as well as their biocompatibility, biodegradability, non-toxic and non-immunogenicity nature (Mathiyazhakan et al ...
Liposomes are indispensable model membranes and drug carriers. Here, the authors use DNA nanostructures to coat, cluster, and pattern sub-100-nm liposomes, enabling attachment of proteins and ...
The Journal of Liposome Research aims to publish original, high-quality, peer-reviewed research on the topic of liposomes and related systems, lipid-based delivery systems, lipid biology, and both synthetic and physical lipid chemistry. Reviews and commentaries or editorials are generally solicited and are editorially reviewed. The Journal also publishes abstracts and conference proceedings ...
The various other drug delivery devices include niosomes, microparticles, resealed erythrocytes, pharmacosomes etc.The term liposome means lipid body. It has been derived on the basis of name of ...
Liposomes are now considered the most commonly used nanocarriers for various potentially active hydrophobic and hydrophilic molecules due to their high biocompatibility, biodegradability, and low immunogenicity. ... 1 Pharmacological and Diagnostic Research Center, Faculty of Pharmacy, Al-Ahliyya Amman University, Amman, 19328, Jordan.
Review article Liposomes: structure, composition, types, and clinical applications☆ Hamdi Nsairata, Dima Khaterb, Usama Sayedc, Fadwa Odehd, Abeer Al Bawabd,e, Walhan Alshaerf,* a Pharmacological and Diagnostic Research Center, Faculty of Pharmacy, Al-Ahliyya Amman University, Amman, 19328, Jordan b Department of Chemistry, Faculty of Arts and Science, Applied Science Private University ...
Lipid nanoparticles (LNPs) have emerged across the pharmaceutical industry as promising vehicles to deliver a variety of therapeutics. Currently in the spotlight as vital components of the COVID-19 mRNA vaccines, LNPs play a key role in effectively protecting and transporting mRNA to cells. Liposomes, an early version of LNPs, are a versatile nanomedicine delivery platform. A number of ...
This review article examines the preparation of modified liposomes using methods such as the thin-film method, reverse evaporation method and injection method. The purpose of liposome modification and its application in food are discussed, with a focus on enhancing targeting, prolonging in vivo circulation time, improving stability and ensuring ...
Research Article. Open Access. Liposomes as Carriers of Membrane-Associated Proteins and Peptides for Mass Spectrometric Analysis. Melissa Frick, Melissa Frick. Interdisciplinary Research Center HALOmem, Charles Tanford Protein Center, Institute for Biochemistry and Biotechnology, Martin Luther University Halle-Wittenberg, Kurt-Mothes-Strasse ...
Systematic evaluation using multiple vials and repeated test runs of three liposomes and three LNP-siRNA formulations showed no toxicity in HL60 and A549 cells up to 128 and 16 µg/mL ...
In this paper, the preparation and characterization of liposomes are briefly summarized, and the research and application of liposomes in organic and biological substances analysis are mainly ...
Abstract. Liposomes, sphere-shaped vesicles consisting of one or more phospholipid bilayers, were first described in the mid-. 60s. Today, they are a very useful reproduction, rea gent, and tool ...
Metrics. Liposomes are ubiquitous components of skin moisturizers and other personal-care products. Modified liposomes prepared from receptor-like molecules open up fresh opportunities for ...
Research on liposome technology has progressed from conventional vesicles to 'second-generation liposomes', in which long-circulating liposomes are obtained by modulating the lipid composition, size, and charge of the vesicle. Liposomes with modified surfaces have also been developed using several molecules, such as glycolipids or sialic ...
Aceclofenac (ACE) is a drug that was precisely devised to circumvent the shortcomings associated with diclofenac. However, ACE too corresponds to nonsteroidal anti-inflammatory drug (NSAID)-related adverse effects, but with a lower amplitude. The present investigation seeks to develop liposomes loaded with ACE adopting a central composite design (CCD) and formulate a chitosan-based hydrogel ...
In an alternative approach to deplete recruited lung monocytes-macrophages induced by environmental exposures, intravenous clodronate liposomes (vs. vehicle control liposomes) were dosed one day prior to LPS (and saline) treatment to reduce circulating/systemic reservoir of available monocytes-macrophages with mice euthanized at 48 h following ...
Nutlin-3a was effectively encapsulated in liposomes and was able to induce a significant reduction of viability and migration in RPE cell models. Conclusion: Our results lay the basis for a possible use of liposomes for the ocular delivery of Nutlin-3a. Keywords: Nutlin-3a, liposomes, microfluidic, PVDs, CFFF.